降脂方中活性成分含量測定方法*
2021-11-24 08:38:36汪怡,劉菊,楊菊
中醫藥導報 2021年11期
汪 怡,劉 菊,楊 菊
(昆山市中醫醫院,江蘇 昆山 215300)
20世紀80年代以來,隨著社會經濟的發展和居民生活方式、膳食結構的改變,中國人群血脂異常患病率明顯增加。中醫藥治療高脂血癥療效顯著,但目前治療方案中,中藥湯劑煎煮步驟繁瑣。近年來的研究表明降脂茶在輔助治療血脂異常方面具有一定作用,并且相比藥物具有更小的毒副作用[1]。
降脂方為我院臨床常用于治療高脂血癥的茶飲基礎方,以茶飲代替湯劑煎煮,簡單方便。本方由荷葉、山楂、炒決明子、澤瀉、丹參、槐米六味藥組成,全方質輕性薄,寒溫并用,清上通下,分消走瀉,氣血水同調,共奏行氣活血、利濕化痰之功,旨在調節血脂代謝,改善高脂血癥臨床癥狀和血脂指標。荷葉和決明子是方中降脂的主要藥物之一,現代藥理學證明,荷葉中的生物堿是降低高脂血癥大鼠血脂和體質量的主要有效成分[2];決明子提取物能明顯降低高血脂動物血清低密度脂蛋白膽固醇和甘油三酯的含量,增加血清高密度脂蛋白膽固醇水平,降血脂成分主要為苷類、多糖、蛋白質及蒽醌類等成分[3]。本文建立了HPLC法同時測定降脂方中橙黃決明素、黃決明素、決明素、決明蒽醌4種蒽醌類成分含量的方法和用酸性染料比色法測定總生物堿,擬為降脂方的質量控制提供依據,同時也為本課題進一步研究降脂方沖泡條件奠定實驗基礎。
1 儀器與試藥
1.1 儀器 LC-20AD高效液相色譜儀(含SPD-20A紫外檢測器和LabSolutions工作站,日本島津儀器有限公司);UV-2600紫外可見分光光度計(日本島津公司);BT 25S電子天平(d=0.01 mg,賽多利斯科學儀器公司);MS3圓周振蕩器(德國IKA公司);KQ2200E超聲波清洗器(昆山市超聲儀器有限公司);HWS-24型電熱恒溫水浴鍋(上海一恒科學儀器有限公司)。
1.2 藥物與試劑 荷葉(批號:181205003)、決明子(批號:190303003)、山楂(批號:190119005)、丹參(批號:190126005)、澤瀉(批號:181228005)、槐米(批號:190311003)均來自蘇州市天靈中藥飲片有限公司,由我院藥學部中藥房副主任中藥師按照2015年版《中華人民共和國藥典》一部鑒定為合格飲片。橙黃決明素對照品(批號:111900-201605,含量98.3%,中國食品藥品檢定研究院);黃決明素對照品(批號:MUST-19111312,純度:99.79%)、決明素對照品(批號:MUST-19060107,純度:99.01%)、決明蒽醌對照品(批號:MUST-19070608,純度:99.62%)、荷葉堿對照品(批號:MUST-19032204,純度:98.91%)均購自成都曼思特生物科技有限公司;乙腈(色譜純,德國Merker公司);溴甲酚綠指示劑(上海潤捷化學試劑公司,批號:20180610);超純水及純水(美國Millipore公司);其他試劑為分析純。
2 方法與結果
2.1 降脂方茶湯溶液的制備 根據降脂方處方用量比例,分別稱取荷葉、山楂、決明子、丹參、澤瀉、槐米飲片,共24 g,放入無紡布茶包,加入剛煮沸的純水500 mL沖泡3次,每次放置30 min后,倒出茶湯,合并3次茶湯,過200目篩后,轉移至2 000 mL量瓶,純水定容。
2.2 對照品溶液的制備
2.2.1 蒽醌混合對照品溶液的制備 精密稱取橙黃決明素、黃決明素、決明素、決明蒽醌對照品適量,置于100 mL量瓶中,甲醇溶解,定容,配置成含橙黃決明素120.90 μg/mL、黃決明素62.90 μg/mL、決明素42.20 μg/mL、決明蒽醌55.20 μg/mL的混合對照品儲備液。再分別精密量取儲備液0.3、0.6、0.9、1.2、1.5、1.8 mL轉移至25 mL量瓶中,加甲醇稀釋至刻度,搖勻,用0.22 μm微孔濾膜濾過,取續濾液,即得。
2.2.2 生物堿對照品溶液的制備 精密稱取荷葉堿對照品適量,置于25 mL量瓶中,甲醇溶解,定容,配置成含荷葉堿0.192 mg/mL的對照品溶液。
2.3 供試品溶液的制備
2.3.1 蒽醌類成分含量測定供試品溶液的制備 精密量取50 mL茶湯溶液,加鹽酸5 mL置水浴中加熱水解30 min,立即冷卻,用三氯甲烷振搖提取4次,每次50 mL,合并三氯甲烷液,回收溶劑至干,殘渣用甲醇使溶解,轉移至25 mL量瓶中,并稀釋至刻度,搖勻,用0.22 μm微孔濾膜濾過,取續濾液,即得。
2.3.2 總生物堿含量測定供試品溶液的制備 取適量茶湯溶液,4 500 r/min離心10 min,分離上層澄清液,即得。
2.4 蒽醌類成分含量測定方法的建立
2.4.1 色譜條件 采用Agilent Eclipse XDB-C18色譜柱(4.6 mm×250 mm,5 μm),流動相:乙腈-0.1%磷酸溶液(40∶60);流速:1 mL/min;檢測波長:284 nm;柱溫:40 ℃;進樣量:20 μL。
2.4.2 檢測波長的選擇 分別精密稱取橙黃決明素、黃決明素、決明素、決明蒽醌對照品適量,分別置于25 mL量瓶中,甲醇溶解,定容,以相應的試劑為空白,采用紫外-可見分光光度法,分別在200~800 nm區間進行全波長掃描。結果見圖1。
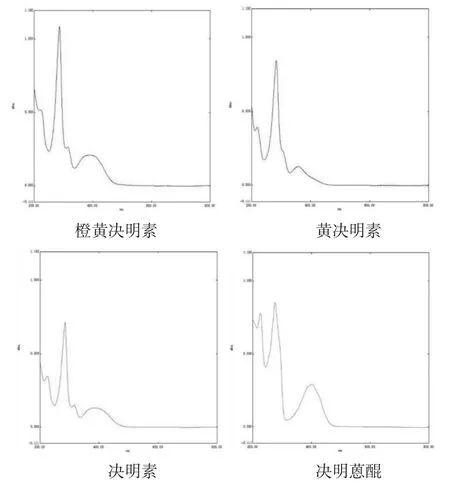
圖1 對照品溶液全波長掃描圖
利用軟件中“峰值檢測”,可知橙黃決明素最大吸收峰為286 nm,黃決明素為283 nm,決明素為284.5 nm,決明蒽醌為276 nm,結合2015年版《中華人民共和國藥典》一部決明子項下含量測定的檢測波長為284 nm,最終選取284 nm為檢測波長。
2.4.3 專屬性考察 陰性對照品溶液的制備:根據處方用量比例,稱取除決明子外的其他飲片制成茶湯溶液,按“2.3.1”項下方法制備陰性對照品溶液。
分別精密吸取混合對照品溶液、供試品溶液、陰性對照品溶液各20 μL,按“2.4.1”項下色譜條件進行測定,結果見圖2,結果表明,供試品色譜圖中,與對照品相同保留時間處有相應色譜峰,陰性對照品色譜圖中,與對照品相同保留時間處無相應色譜峰,陰性無干擾。
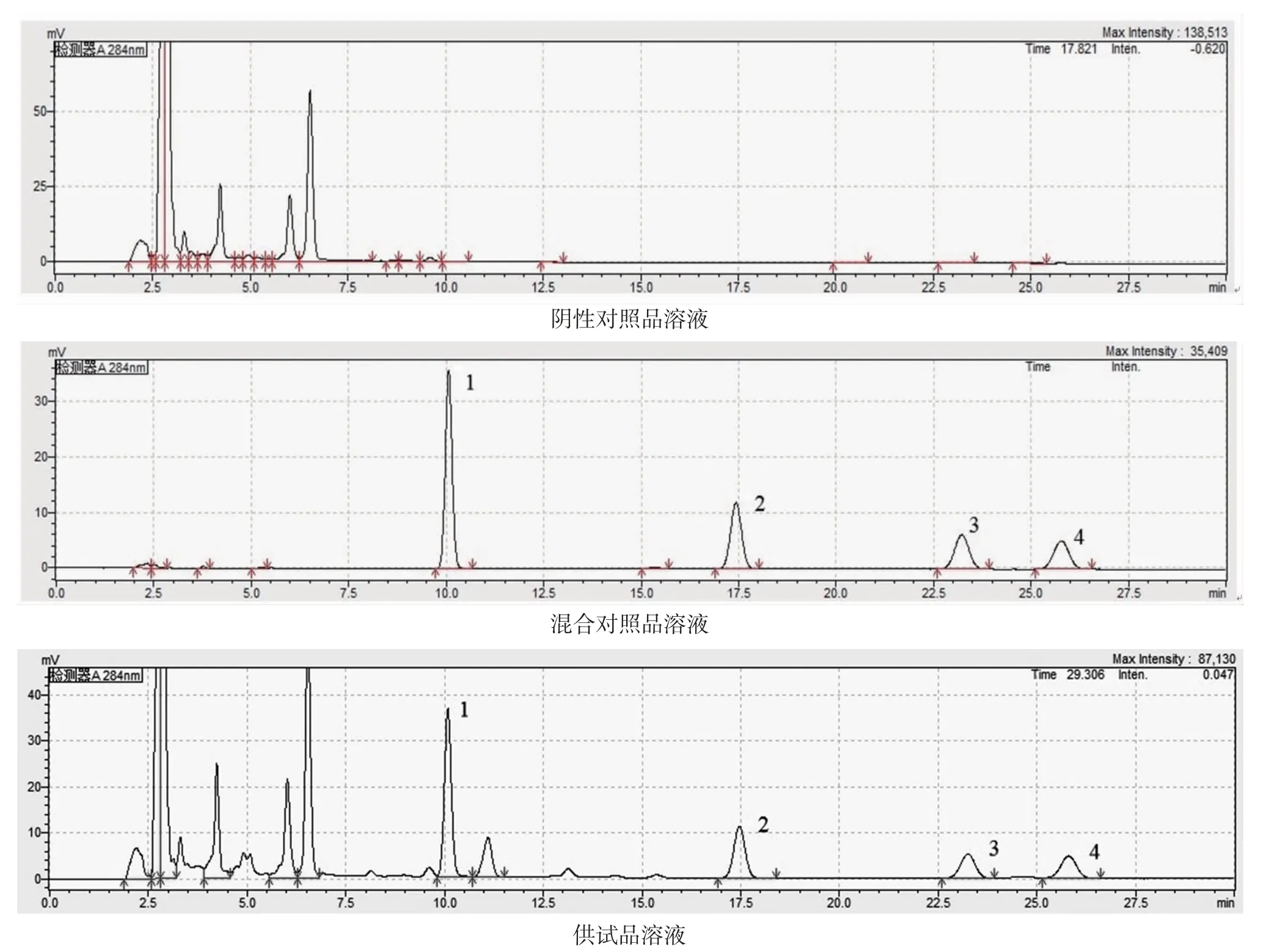
圖2 各樣品溶液HPLC 圖
2.4.4 線性關系考察 分別精密吸取“2.2.1”項下混合對照品溶液20 μL,按“2.4.1”項下色譜條件進行測定,記錄色譜峰峰面積,分別以進樣量(μg)為橫坐標(X),峰面積為縱坐標(Y)繪制標準曲線并進行線性回歸分析,得各對照品的標準曲線回歸方程、線性范圍和相關系數(見表1),結果表明,各組分在相應線性范圍內,峰面積與濃度具有良好的線性關系。
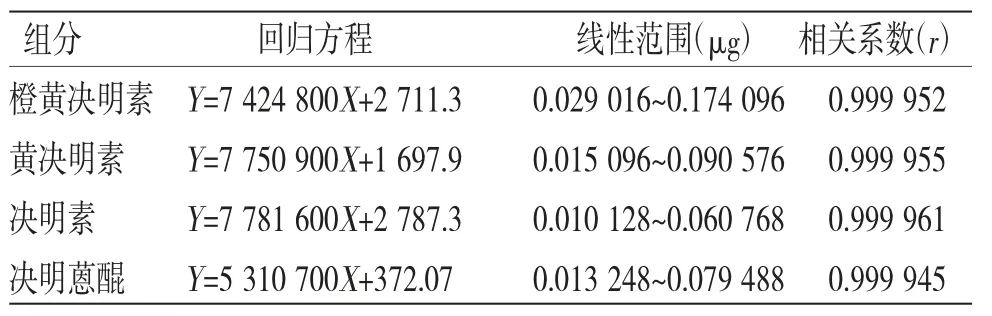
表1 線性關系考察結果
2.4.5 精密度試驗 精密吸取“2.2.1”項下同一混合對照品溶液20 μL,按“2.4.1”項下色譜條件進行測定,連續進樣6次,記錄色譜峰峰面積,計算橙黃決明素、黃決明素、決明素和決明蒽醌峰面積的RSD值分別為1.50%、1.54%、1.52%、1.58%。結果表明儀器精密度良好。
2.4.6 穩定性試驗 精密吸取同一供試品溶液20 μL,分別在0、2、4、6、12、24 h按“2.4.1”項下色譜條件進行測定,記錄色譜峰峰面積,計算橙黃決明素、黃決明素、決明素和決明蒽醌峰面積的RSD值分別為1.56%、1.77%、1.50%、1.52%。結果表明供試品溶液各組分在24 h內穩定。
2.4.7 重復性試驗 取同一茶湯溶液6份,按“2.3.1”項下方法,制備供試品溶液,精密吸取供試品溶液20 μL,按“2.4.1”項下色譜條件進行測定,記錄色譜峰峰面積,計算橙黃決明素、黃決明素、決明素和決明蒽醌在茶湯溶液中的平均含量和RSD,結果見表2。結果表明本方法重復性良好。

表2 重復性試驗結果
2.4.8 加樣回收率試驗 精密量取已知含量的茶湯溶液6份,每份25 mL,分別精密加入同一濃度混合對照品溶液25 mL(精密量取混合對照品儲配液3.5 mL,轉移至250 mL量瓶中,加水稀釋至刻度,搖勻),按“2.3.1”項下“加鹽酸5 mL”起,制備供試品溶液,精密吸取各供試品溶液20 μL,按“2.4.1”項下色譜條件進行測定,記錄色譜峰峰面積,計算加樣回收率和RSD,結果見表3。結果表明本方法的準確度符合要求。

表3 加樣回收率試驗結果
2.4.9 樣品含量測定 分別制備3份茶湯溶液,按“2.3.1”項下方法,制備供試品溶液,精密吸取各供試品溶液20 μL,按“2.4.1”項下色譜條件進行測定,記錄色譜峰峰面積,計算橙黃決明素、黃決明素、決明素和決明蒽醌在茶湯溶液中含量及藥材中含量,結果見表4。

表4 樣品含量測定結果(n=3)
2.5 總生物堿含量測定方法的建立
2.5.1 溴甲酚綠溶液的制備 取溴甲酚綠0.1 g,加0.05 mol/L氫氧化鈉溶液2.8 mL,再加水稀釋至200 mL,即得。
2.5.2 反應及測定條件的選擇
2.5.2.1 緩沖溶液pH值選擇 分別配制pH值為3.2、3.6、4.0、4.4、4.8、5.2、5.4、6.0的檸檬酸-檸檬酸鈉緩沖溶液。
精密吸取供試品溶液10.0 mL數份,置具塞試管中,分別精密加入不同pH值的檸檬酸-檸檬酸鈉緩沖溶液5.0 mL,隨后加入溴甲酚綠溶液2.0 mL,搖勻,再加入二氯甲烷10.0 mL,置于渦旋振蕩器振搖20 s,轉移至分液漏斗中,靜止15 min,分取二氯甲烷層(放入無水硫酸鈉0.5 g),以二氯甲烷為參比,在200~600 nm處分別進行全波長掃描,結果見圖3。
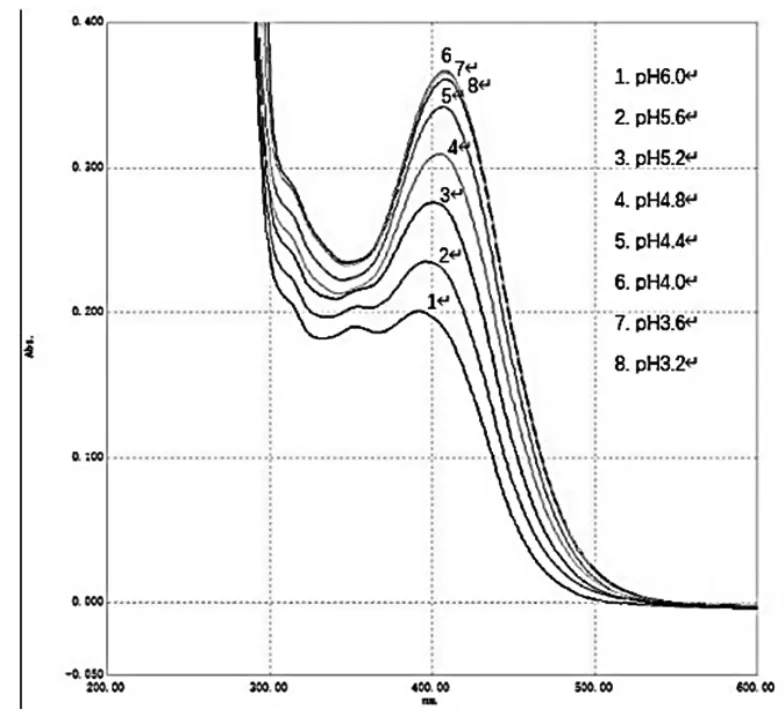
圖3 不同pH 緩沖溶液條件下樣品溶液全波長掃描圖
根據實驗結果分析,在不同pH值緩沖溶液條件下,各樣品溶液在400 nm左右均有特征吸收峰,吸收峰的最大吸收波長(λmax)和最大吸光度(Amax)均有所不同(見表5),選擇吸收峰吸光度最大的pH值緩沖溶液作為實驗溶液,故最終選擇pH值為4.0緩沖溶液。
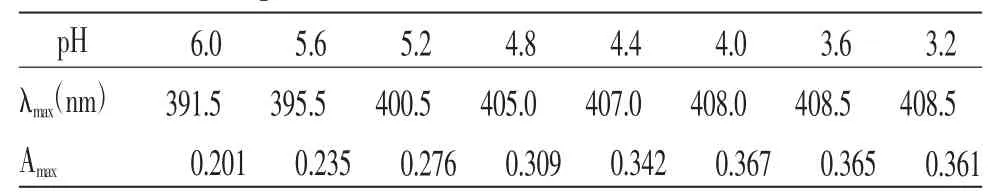
表5 不同pH 緩沖溶液條件下樣品溶液的最大吸收
2.5.2.2 測定波長的選擇 精密吸取對照品溶液0.8 mL,于25 mL容量瓶中,加水稀釋并定容至刻度。精密吸取此對照品溶液10.0 mL,置具塞試管中,精密加入pH值為4.0的檸檬酸-檸檬酸鈉緩沖溶液5.0 mL,隨后加入溴甲酚綠溶液2.0 mL,搖勻,再加入二氯甲烷10.0 mL,置于渦旋振蕩器振搖20 s,轉移至分液漏斗中,靜止15 min,分取二氯甲烷層(放入無水硫酸鈉0.5g),以二氯甲烷為參比,在200~600 nm處進行全波長掃描。根據實驗結果,對照品溶液的最大吸收峰為410 nm,故選擇410 nm為測定波長。
2.5.2.3 酸性染料用量選擇 精密吸取供試品溶液10.0 mL 5份,置具塞試管中,精密加入pH值為4.0的檸檬酸-檸檬酸鈉緩沖溶液5.0 mL,隨后分別加入溴甲酚綠溶液1.0、2.0、3.0、4.0、5.0 mL,搖勻,再加入二氯甲烷10.0 mL,置于渦旋振蕩器振搖20 s,轉移至分液漏斗中,靜止15 min,分取二氯甲烷層(放入無水硫酸鈉0.5 g),以二氯甲烷為參比,于410 nm處測定吸光度。結果見表6。結果表明酸性染料用量選擇3.0 mL。
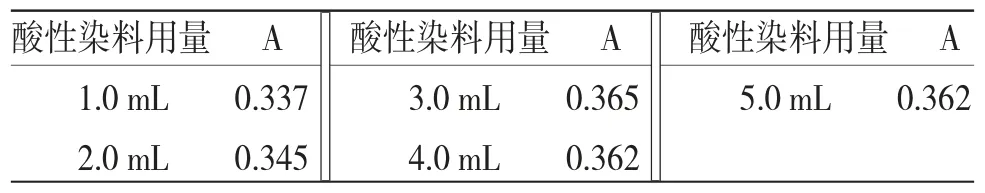
表6 酸性染料用量選擇實驗結果
2.5.2.4 緩沖溶液用量的選擇 精密吸取供試品溶液10.0 mL 5份,置具塞試管中,分別精密加入pH值為4.0的檸檬酸-檸檬酸鈉緩沖溶液2.0、4.0、6.0、8.0、10.0 mL,隨后加入溴甲酚綠溶液3.0 mL,搖勻,再加入二氯甲烷10.0 mL,置于渦旋振蕩器振搖20 s,轉移至分液漏斗中,靜止15 min,分取二氯甲烷層(放入無水硫酸鈉0.5 g),以二氯甲烷為參比,于410 nm處測定吸光度。結果見表7。結果表明緩沖溶液用量選擇2.0 mL。

表7 緩沖溶液用量選擇實驗結果
2.5.3 線性關系考察 精密吸取對照品溶液0.4、0.6、0.8、1.0、1.2、1.4 mL,于25 mL容量瓶中,加水稀釋并定容至刻度。精密吸取以上各對照品溶液10.0 mL,置具塞試管中,精密加入pH值為4.0的檸檬酸-檸檬酸鈉緩沖溶液2.0 mL,隨后加入溴甲酚綠溶液3.0 mL,搖勻,再加入二氯甲烷10.0 mL,置于渦旋振蕩器振搖20 s,轉移至分液漏斗中,靜止15 min,分取二氯甲烷層(放入無水硫酸鈉0.5 g),以二氯甲烷為參比,照紫外可見分光光度法(2015年版《中華人民共和國藥典》四部附錄0401),于410 nm波長處測定吸光值。以吸光度為縱坐標,濃度為橫坐標,進行線性回歸。得到以下線性回歸方程Y=0.006 8X-0.0144,r=0.9998,結果表明當荷葉堿對照品在30.72~107.52 μg范圍內與吸光度有良好的線性關系。
2.5.4 精密度試驗 精密吸取對照品溶液3.2 mL,于100 mL容量瓶中,加水稀釋并定容至刻度。再精密吸取此對照品溶液10.0 mL,6份,按“2.3.1”項下“置具塞試管中”起,依法測定吸光度,RSD為1.30%,結果表明該方法精密度較好。
2.5.5 穩定性試驗 精密吸取供試品溶液10.0 mL,按“2.3.1”項下“置具塞試管中”起,依法每隔30 min,共180 min內測定吸光度,RSD為0.20%,結果表明該方法在180min內穩定性良好。
2.5.6 重復性試驗 按“2.1.2”項下供試品溶液制備方法同一樣品溶液制備6份,精密吸取各供試品溶液10.0 mL,按“2.3.1”項下“置具塞試管中”起,依法測定吸光度。計算得平均含量以荷葉堿計算為5.871 μg/mL,RSD為1.97%。結果表明方法的重復性較好。
2.5.7 加樣回收率試驗 精密吸取已知含量供試品溶液5.0 mL 6份,置于具塞試管中,隨后分別精密加入同一濃度對照品溶液5.0 mL,按“2.3.1”項下“置具塞試管中”后起,依法測定吸光度,計算回收率。平均回收率為102.10%,RSD為1.91%。(見表8)

表8 加樣回收率實驗結果
2.5.8 含量測定 按上述方法,測定3批降脂方樣品溶液中總生物堿含量,結果其生藥中總生物堿含量以荷葉堿計算分別為0.492、0.476、0.425 mg/g。
3 討 論
3.1 蒽醌含量測定方法 降脂方中蒽醌類成分主要來源于決明子,主要為大黃素型蒽醌,呈游離或結合狀態,樣品需經過水解將結合態轉變成游離態進行測定,結合2015年版《中華人民共和國藥典》一部決明子[4]項下含量測定的供試品溶液的制備,利用正交實驗,選用L9(34)正交表,考察鹽酸用量(50 mL茶湯溶液分別加入5、15、25 mL鹽酸)、水解時間(水浴加熱30、75、120 min)、振搖提取次數(50 mL三氯甲烷分別振搖提取2次、3次、4次)三因素三水平的影響情況,方差分析結果顯示,各因素水平對結果影響并不顯著,最終根據直觀分析,選擇水解條件為加鹽酸5 mL置水浴中加熱水解30 min,立即冷卻,用三氯甲烷振搖提取4次,每次50 mL。
在確定檢測指標成分和流動相時,曾參照2015年版《中華人民共和國藥典》一部決明子[4]項下含量測定的流動相條件以及文獻資料[5-8]進行試驗,色譜圖中對應有多個蒽醌類成分色譜峰,按保留時間為橙黃決明素、黃決明素、決明素、決明蒽醌、大黃素、大黃酚、大黃素甲醚,但由于大黃素、大黃酚、大黃素甲醚在茶飲沖泡條件下含量較低,故選擇橙黃決明素、黃決明素、決明素、決明蒽醌4個含量相對較高的成分作為檢測指標成分,同時調整流動相條件,最終選擇乙腈-0.1%磷酸溶液(40∶60)等度洗脫,使各指標成分峰之間能達到較好的基線分離。
3.2 總生物堿含量測定方法 目前已知的荷葉中分離得到的生物堿,按照生源途徑結合化學結構類型分類,可將它們歸為苯丙氨酸和酪氨酸系生物堿中的異喹啉類[2],而在萜類、吖啶酮類、甾類、有機胺類、異喹啉類或多種類型混合生物堿中,溴甲酚綠和溴麝香草酚藍均有應用,其中異喹啉類生物堿應用溴甲酚綠較多[9]。故本實驗的酸性染料選擇用溴甲酚綠。
酸性染料比色法的原理是在適宜酸性條件下,生物堿陽離子可與染料陰離子定量結合,生成水難溶性有色離子對產物,有機溶劑萃取后,可以在紫外可見分光光度計中測定有機層溶液的吸光度,根據標準曲線計算出生物堿的含量。因此,緩沖溶液的種類、緩沖溶液pH和用量、酸性染料用量等都會對反應產生影響。本實驗對這些影響因素進行了比較詳細的研究,確定了最佳反應條件[10-17]。
在比色過程中,水分的混入會影響測定結果,因為水相中有過量的有色染料,同時水的混入使有機溶劑混濁,影響比色[18],因此加入了一定量的無水硫酸鈉作為脫水劑。本實驗還比較了二氯甲烷和三氯甲烷的萃取效果,發現用二氯甲烷萃取所測的吸光度更大,且二氯甲烷毒性較低,故實驗選用二氯甲烷進行萃取。
綜上所述,本實驗建立了HPLC法同時測定降脂方中橙黃決明素、黃決明素、決明素、決明蒽醌這4種蒽醌類成分含量的分析方法和利用酸性染料比色法測定降脂方中總生物堿的方法。方法操作簡便,結果準確可靠,專屬性強,重復性好,為本課題今后進一步研究降脂方沖泡條件奠定了實驗基礎。
