拉伸形變及電場作用對黑磷烯吸附Si 原子電學特性影響的密度泛函理論研究*
2021-11-19 05:16:08衛琳劉貴立王家鑫穆光耀張國英
物理學報 2021年21期
衛琳 劉貴立? 王家鑫 穆光耀 張國英
1) (沈陽工業大學建筑與土木工程學院,沈陽 110870)
2) (沈陽師范大學物理學院,沈陽 110034)
構建了覆蓋度為2.778%的黑磷烯吸附硅原子模型,基于密度泛函理論計算了模型的電子特性,并通過應力及電場對其電子特性進行調控.研究表明:當前研究的覆蓋度下,Si 原子的吸附導致黑磷烯幾何對稱性被破壞,加劇了體系內的電荷轉移,完成軌道再雜化.使黑磷烯帶隙消失,實現了其由半導體向準金屬的轉變.其穩定的吸附位是位于P 原子環中間的H 位.拉伸和電場均降低了黑磷烯體系穩定性.拉伸形變使黑磷烯吸附Si 原子結構打開帶隙,且帶隙與形變量成正比,實現對其帶隙的調控.電場與拉伸共作用下,電場的引入使黑磷烯吸附Si 原子帶隙變窄且完成體系由直接帶隙向間接帶隙的轉變.帶隙依舊隨形變量增加而增加.吸附Si 原子的黑磷烯體系帶隙可調性高于未吸附體系,且易于實現帶隙的穩定調控.
1 引言
黑磷烯是一種二維層狀半導體材料,它具有高載流子遷移率、高開/關比、強各向異性等性質[1-12],在熱電、光電、能量儲存、傳感器和場效晶體管等領域有著廣泛的應用前景.
從原子層面講,硅的化合價半徑類似于磷的化合價半徑,且兩原子相對原子質量接近,使得兩者之間具有很多聯系.黑磷烯作為磷單質的二維材料,與硅這種元素相結合亦有許多應用.首先是有關黑磷烯的制備方面.從由最初的機械剝離方法[13,14]到近期報道的一種在硅基底上生長高結晶性黑磷薄膜的新方法,黑磷烯在生產制備方面取得了極大地進展.Rajabali 等[14,15]通過激光激發在硅襯底上形成高度結晶的薄片來形成磷烯.Xu等[16]通過氣相生長策略在絕緣硅襯底上直接生長大規模晶體黑磷烯薄膜.這種直接在硅襯底上生長的黑磷烯具有層數少、制造面積大、穩定性強、可調性強等優點.
其次是黑磷烯在等離子體方面的應用[17,18].Prajapati 等[19]利用硅-黑磷-TDMC 涂層表面等離子體共振生物傳感器提高靈敏度.Kumar 等[20]探討了硅混合黑磷納米結構表面等離子共振生物傳感器的靈敏度.Kumar 等[21]采用五層黑磷-硅基隧道場效應晶體管克服熱離子限制.
再次是黑磷烯在硅基電池方面的應用.硅基材料用作陽極的硅基鋰離子電池在使用和制造過程中存在一些弊端,如材料粉碎、活性材料晶粒間導電損失[22,23]、活性物質損失等[22,24].這些缺陷制約了硅基鋰離子電池的發展.而黑磷烯由于其特殊性能[25-28],在能量存儲器件中具有應用前景.Zhang等[27]報道了鋰離子在磷烯中的擴散速度很快,在硅陽極中引入磷烯有望促進鋰離子的吸收.Park和Sohn[29]的研究表明多層黑磷-碳復合材料用作電池表現出優異的循環性能.一些研究者[30-34]將磷摻雜到作為硅基電池材料的硅晶格中進行研究,結果表明磷是改善鋰離子電池中硅陽極電化學性能的有效方法.Peng 等[35]從石墨烯等納米結構碳質材料表面封裝中得到啟發,使用單層黑磷烯包覆硅顆粒來提升硅基負極材料的電化學性能.
由此可見,磷烯與硅相結合的應用較為廣泛,在硅基底上生長黑磷烯時,襯底硅對其電子性質有著不可忽視的影響.當黑磷用作等離子體傳感器及晶體管時,硅層和黑磷層直接接觸,其層間影響對傳感器靈敏度和場效應晶體管影響較大.在黑磷烯用于包覆硅基鋰離子電池陽極材料硅時,兩者相互接觸.所以研究硅原子與黑磷之間的相互作用十分必要.目前,使用第一性原理方法對磷烯與硅之間的研究僅局限于摻雜方面,Shojaei 等[36]研究了硅摻雜單層和雙層磷烯納米帶中的硅-硅鍵效應.結果表明:雙層磷烯納米帶摻雜硅后,結構都變成了間接間隙半導體.Olmedo 等[37]研究了硅摻雜的磷烯納米片物性,結果表明,摻雜使幾何結構改變,磷烯納米片帶隙減小.但以上研究均基于實驗方法,側重黑磷烯制備及應用方面,并非直接探尋黑磷烯與硅原子之間的相互作用.僅有的幾個理論模擬也均為硅原子摻雜黑磷烯物性研究,而層間接觸、包覆材料等則需要通過吸附特性進行深入研究.目前,有關黑磷烯吸附Si 原子的研究未見報道.所以本文基于第一性原理研究黑磷烯吸附硅原子的電子特性,并期望對其電子特性實現穩定調控.希望該研究對于硅電池、半導體器件、等離子體生物傳感器有指導作用.
2 理論模型與計算方法
構筑單層黑磷烯材料,研究吸附Si 原子對其電子特性的影響.通過對黑磷單胞進行修剪得到二維黑磷烯的單晶結構,其原胞中含有4 個磷原子,晶體結構屬于正交晶系 (orthorhombic),空間族群為CMCA (No.64),如圖1(a)中的藍色區域所示,其中a和b代表晶格常數.將原胞沿黑磷烯面內基矢a和b方向擴展3 倍,建立3 × 3 × 1 的單層黑磷超胞,用來表示黑磷烯平面.在垂直單層黑磷超胞的晶面方向上設置15 ?的真空層以消除相鄰層間的相互作用,模型主視圖、俯視圖、側視圖和晶胞結構示意圖如圖1 所示.圖中Δc表示單層黑磷的垂直層內間距,d1和d2為所標記的P—P 鍵鍵長,α與β為黑磷原子間鍵角.文中所有密度泛函理論計算均采用Material Studio (MS)軟件中的CASTEP 模塊[38]進行.采用GGA-PBE 泛函進行處理[39],由于廣義梯度近似(GGA)低估了帶隙,因此帶結構采用雜化泛函Heyd-Scuseria-Ernzerhof(HSE06)[40]進行計算.黑磷烯超胞的整體能量收斂性測試過程中,平面波截斷能設置為400 eV[41-43],布里淵區K點網格設置為3 × 3 × 3.幾何優化參數設置為:自洽場迭代收斂判據為2.0 × 10—6eV;結構優化的收斂判據為0.02 eV/?.
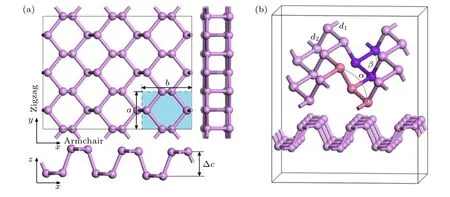
圖1 本征黑磷烯模型 (a)黑磷烯的主視圖、俯視圖、側視圖;(b)黑磷烯結構示意圖Fig.1.Intrinsic black phosphorene model:(a) Front view,top view,and side view of black phosphorene;(b) schematic diagram of the structure of black phosphorene.
首先對黑磷烯原胞進行幾何結構優化,結構優化后其晶格常數a=3.313 ?,b=4.374 ?,與文獻[44-48]相符.然后對擴展后的黑磷烯超胞進行結構優化,優化后的黑磷烯片層厚度Δc=2.093 ?,與文獻[46]的模擬結果2.110 ?相比誤差為0.081%;P—P 原子的鍵長d1=2.210 ?,d2=2.230 ?,與文獻[44,48]的結果相符;鍵角α=103.016°,β=98.539°與文獻[48,49]的模擬結果非常接近,∠α與文獻[50]所得的角度102.31°相比誤差為0.69%.說明本文參數設置合理.
形變和電場會影響材料的穩定性,通常較大的形變會導致穩定性降低,單原子結合能的定義式[51-55]為

其中EBP為黑磷的總能量,Ep為單個磷原子的能量,np為晶胞中磷原子的數目.結合能為負值表明原子從孤立狀態變成結合態會放出能量,放出的能量越大,結合物越穩定.若結合能為正值,該物質不能穩定存在.
此時黑磷吸附體系的吸附能Ead[56]可以表示為
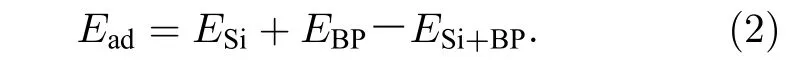
式中,ESi為單個Si 原子的能量,EBP為黑磷烯體系(包括本征、拉伸、電場作用的黑磷烯體系)的能量,ESi+BP為吸附Si 原子后體系的能量.吸附能越大吸附越穩定,若吸附能為負,則不能吸附.
3 計算結果與分析
3.1 本征黑磷烯電子特性
根據(1)式得到的單原子結合能為—5.774 eV.計算黑磷電子態密度時,布里淵區積分采用Monkhorst-Pack 特殊K點對全布里淵區求和,能帶計算高對稱K點路徑為Γ(0,0,0)→Y(0,0.5,0)→L(0.5,0.5,0)→X(0.5,0,0) →Γ(0,0,0).從圖2可以看出,純單層黑磷晶體價帶頂(VBM)出現在Γ點,導帶底(CBM)非常靠近Γ點.體現為近直接帶隙半導體,這點與文獻[57,58]得到的結論相同.其帶隙顯示為1.578 eV,與文獻[48,59-63]相同.態密度(DOS)圖表明黑磷烯在導帶內及價帶高能級部分能級主要由P 原子的p 軌道貢獻,而價帶的低能級區主要由P 原子的s 軌道主導.

圖2 黑磷烯能帶結構和DOSFig.2.Band structure and DOS of black phosphorene.
3.2 吸附Si 原子前后黑磷烯體系物性變化
3.2.1 黑磷烯吸附Si 原子模型建立及吸附特性分析
在優化好的黑磷烯超胞結構P 原子層上方吸附Si 原子.考慮圖3 所示的頂位(T)、橋位(B)和空位(H) 3 個吸附位.為了方便表述,將黑磷烯吸附Si 原子超晶胞體系中的磷原子依次編號,如圖3(c)所示.對吸附體系來說,吸附原子的百分比的影響非常重要[64].選取吸附位H,建立了覆蓋度為2.778%,5.556%和8.333%的黑磷烯吸附Si 原子體系結構模型,在結構優化的基礎上進行了電子特性計算.結果表明吸附原子Si 的覆蓋度對于黑磷烯電子特性有著巨大影響,覆蓋度由2.778%增加到5.556%時,帶隙由0 銳增為0.731 eV,之后覆蓋度增加為8.333%時,黑磷吸附體系帶隙又減小到0.216 eV.由于覆蓋度為2.778%的黑磷烯吸附Si 原子模型使帶隙改變較為明顯,引發了其由半導體至準金屬的改變,研究意義較大.所以如未特殊說明,本文接下來的計算均選取覆蓋度為2.778%的黑磷烯吸附Si 原子模型,即超胞內含有1 個Si原子和36 個P 原子.

圖3 黑磷烯吸附Si 原子模型 (a)主視圖;(b)示意圖;(c) P 原子編號示意圖Fig.3.Si adsorbed on black phosphorene model:(a) Main view;(b) diagrammatic sketch;(c) numbering diagram of P atom.
黑磷烯吸附Si 原子幾何優化后的結構如圖4所示.可以看出Si 原子吸附在黑磷烯上后,吸附原子附近的黑磷烯平面略微向下凹陷,對Si 原子形成略微包裹的趨勢.根據(2)式計算了體系的吸附能,T 位吸附能為3.507 eV,B 位為2.751 eV,H 位為3.657 eV,均屬于可逆化學吸附.可知當Si 原子吸附在P—P 原子環中間的H 位時體系能量最小,吸附能最大,表示Si 原子吸附在黑磷烯表面越穩定.將吸附高度d0定義為Si 原子到BP 表面的垂直距離.為表征其吸附特性及成鍵穩定性,將最短P—Si 鍵鍵長,吸附原子Si 所在P 原子環上的4 個P—P 鍵鍵長和吸附高度列于表1.結合圖4 的結構優化圖和表1 可以看出,Si 原子吸附在黑磷烯的B 位上鍵長改變最大,幾何形變最強烈,吸附高度最高.吸附能也表明此結構穩定性最弱.幾何優化后,在黑磷烯上T 位吸附的Si 原子被自動優化到H 位上.兩個模型的鍵長相差不大,幾何結構相似,吸附能相近.因此,后續的計算都將Si 原子吸附在黑磷烯的H 位.
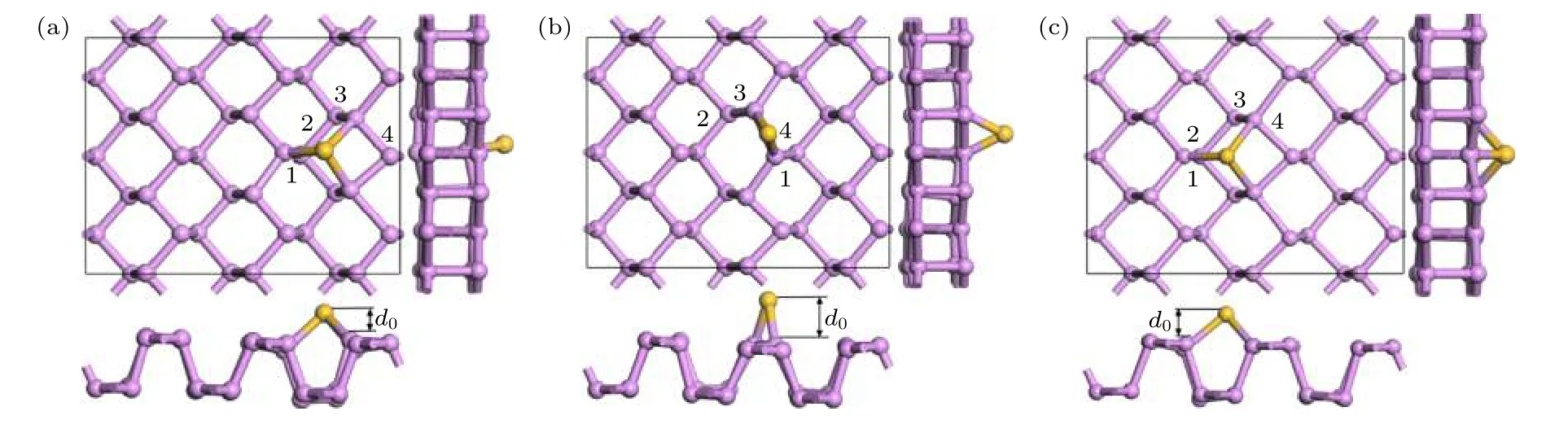
圖4 黑磷烯吸附Si 原子模型幾何優化結構 (a) T 位;(b) B 位;(c) H 位Fig.4.Geometry optimization of Si adsorbed on black phosphorene model:(a) T site;(b) B site;(c) H site.

表1 吸附原子所在原子環的P—P 鍵鍵長與Si 原子吸附高度Table 1.Relationship between P—P bond length and Si adsorption height.
3.2.2 黑磷烯吸附Si 原子電子特性
圖5 和圖6 展示了本征黑磷烯和黑磷烯吸附Si 原子的能帶和態密度結構.可以看出Si 原子吸附在黑磷烯上的T,B 和H 位上時,帶隙分別為0.055,0.072 和0 eV.T 位和B 位吸附的黑磷烯體系帶隙均為間接帶隙且小于0.1 eV,體現出準金屬特性.H 位吸附體系導帶和價帶之間發生重疊,禁帶消失,電子可以無障礙地達到導帶.
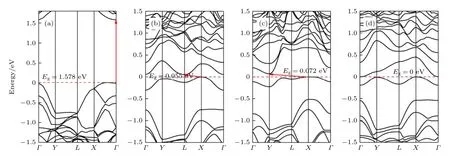
圖5 黑磷體系能帶結構 (a)本征黑磷烯;(b) T 位吸附;(c) B 位吸附;(d) H 位吸附Fig.5.Band structure of black phosphorene system:(a) Intrinsic black phosphorene;(b) T site adsorption;(c) B site adsorption;(d) H site adsorption.
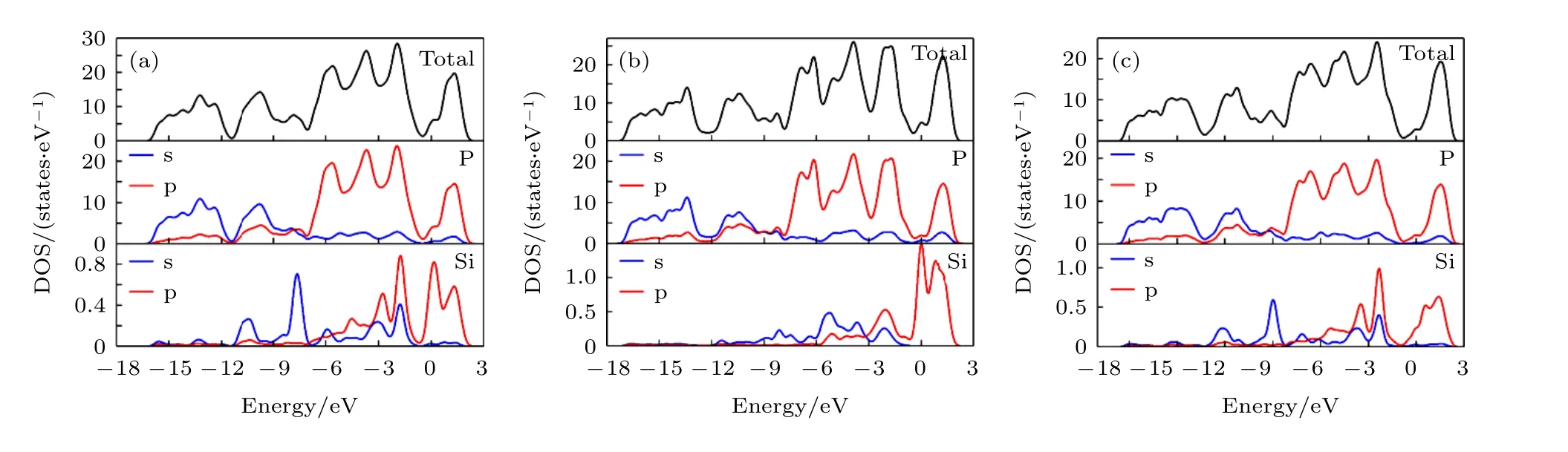
圖6 黑磷烯吸附體系DOS (a) T 位;(b) B 位;(c) H 位Fig.6.The DOS of Si adsorbed on black phosphorene system:(a) T site;(b) B site;(c) H site.
為了探究H 位的Si 吸附使黑磷烯結構禁帶消失的機理,研究了吸附體系每一個P 原子在費米能級附近的DOS 貢獻,并將貢獻較多的原子按照圖例顏色在模型中依次顯示,如圖7 所示.從圖7可以看出,在費米能級處,不完全是Si 原子的貢獻,大部分是黑磷烯本身的貢獻,按照貢獻由大到小排列依次為P10,34> P16,28> Si > P5> P22>P20> P14,26> P4.其余原子貢獻低于0.05 eV,固此處不予討論.P 原子的分波態密度(PDOS)曲線表明,吸附Si 原子后的黑磷烯在結構上仍然具有對稱性,P10,16,14與P34,28,26的DOS 曲線重合.貢獻較高的P 原子集中在吸附原子附近沿zigzag方向的一排P 原子(即圖7 中編號為4,10,16,28和34 的一排磷原子).為了從幾何構型、成鍵、軌道重疊等方面解釋禁帶消失的原因,接下來給出了費米能級處DOS 貢獻較多的P 原子間鍵長、Mulliken 鍵級,所有P 原子的電荷布居數,以及吸附體系的電荷差分密度及電荷密度.

圖7(a) 單個P 原子DOS 圖;(b) P 原子得失電子示意圖;紅色球體代表得到電子的P 原子,藍色球體代表失去電子的P 原子Fig.7.(a) DOS diagram of single P atom;(b) schematic diagram of gain and loss of electrons of P atom.The red sphere represents the P atom that gets electrons,and the blue sphere represents the P atom that loses electrons.
表2 列出了P 原子間鍵長及鍵級.從幾何構型來說,Si 原子的吸附改變了黑磷烯的局域結構,吸附原子附近的P 原子位置及原子間鍵長改變劇烈.吸附破壞了黑磷烯armchair 方向的原始對稱性,但是仍舊保留了其zigzag 方向的部分對陣性.過吸附原子沿x方向做軸線,黑磷烯的上半部分與下半部分幾何形狀完全一致.這點也與DOS 得到的結論一致.正是由于armchair 方向的原始對稱性被破壞,黑磷烯平面發生小范圍形變,P—P 鍵鍵長與鍵角變化,導致磷原子間σ 軌道重疊程度改變.再雜化作用使得其電子特性變化,禁帶消失.

表2 P 原子間鍵長及鍵級Table 2.Bond length and bond order between P atoms.
將吸附體系中磷原子Mulliken 電荷布居數列于表3,圖8 和圖9 顯示了體系的電荷差分密度和電荷密度.結合圖表對電荷轉移及成鍵類型進行分析.從表3、圖8 和圖9 可以看出,Si 原子吸附到黑磷烯表面,失去了0.22e的電子,全都轉移到了黑磷烯上.Si 附近的3 個P 原子P16,28和P20的電荷布居數分別為—0.06e和0.06e,表明它們得到和失去電子的數量為0.06e.通過電荷差分密度圖及電荷密度圖也可證實,P 原子與Si 原子相互之間有強烈的電子遷移,成鍵離子性很強.磷原子中電子改變最為劇烈的原子當屬P10和P34,它們得到的電子數量為0.1e.這點也體現在差分電荷密度和電荷密度圖中,P10—P16和P28—P34之間幾乎無電子轉移,離子性很弱.這可能是由于吸附Si 原子后,P10—P16和P28—P34鍵被拉長,兩原子軌道重疊程度降低,分子軌道重新排布后更多電子聚集在P原子上.它們的鍵級由本征P—P 的0.47 變為1,說明這兩對P 原子成共價鍵.正是由于吸附Si 原子后,Si 原子與黑磷烯間形成離子鍵.強烈的電子轉移更是改變了黑磷烯的電子體系,加劇了黑磷烯本身P 原子間的電荷轉移,改變部分原子成鍵類型.Si 的吸附也在費米能級處引入了一條雜質能級.這可能就是Si 原子使黑磷烯體系能帶結構發生變化,使其由半導體完成向準金屬轉變的原因.

表3 P 原子的Mulliken 電荷布居數Table 3.Mulliken charge population of P atom.

圖8 黑磷烯電荷差分密度圖 (a)本征黑磷烯;(b)黑磷烯吸附Si 原子Fig.8.Differential charge density of the black phosphorene:(a) Intrinsic black phosphorene;(b) Si adsorbed on black phosphorene system.
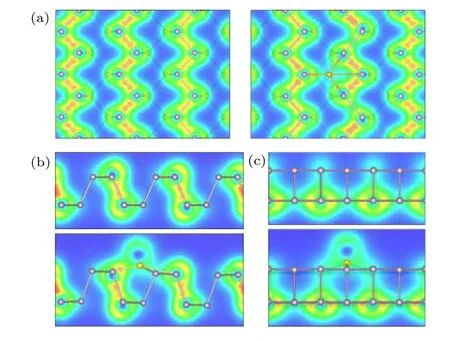
圖9 本征黑磷烯與黑磷烯吸附體系電荷密度圖 (a)主視圖;(b)俯視圖;(c)側視圖Fig.9.Charge density diagram of the adsorption system of intrinsic black phosphorene and black phosphorene:(a) Main view;(b) top view;(c) side view.
3.3 拉伸形變對黑磷烯吸附Si 原子體系影響
3.3.1 黑磷烯拉伸模型建立及結構穩定性分析
眾所周知,黑磷烯具有強各向異性和負泊松比等特性.許多學者的計算結果表明,其電學性能,如帶隙等對外加形變非常敏感[2,8,54,58,60-63,6568],會引起黑磷直接帶隙與間接帶隙的轉變[60,62,63,68],甚至會引起其由半導體到金屬的改變[58].也有許多研究者對黑磷烯進行拉伸和壓縮實驗[69-72].Peng等[68]發現,磷烯可以分別承受高達10 N/m 和30%的拉伸應力和應變.已有研究表明,黑磷烯所受應變在15%以內時,其晶體結構沒有任何明顯損傷[73,74].基于此,對黑磷烯吸附Si 原子體系施加形變,希望實現其帶隙調控.通過改變晶格參數的方式沿armchair 方向對黑磷烯體系施加2%—10%的拉伸形變,所有結構均被充分馳豫.黑磷烯的拉伸形變的程度用形變量ε來表征,將其定義為ε=(c—c0)/c0,式中c0和c分別表示黑磷烯原胞的晶格長度和拉伸后的黑磷烯晶格長度[75].由于黑磷烯具有強各向異性和奇特的負泊松比的現象,充分考慮泊松比的影響,選取拉伸形變量為2 的黑磷烯進行試算.黑磷烯在armchair 和zigzag 兩個方向上的泊松比分別為0.01 和0.16[76],垂直于黑磷烯片層方向上的泊松比為—0.027[77].在對黑磷烯的armchair 方向施加的形變量ε=2%時,在zigzag方向同時施加—0.16Δc=—0.020 ?的形變,垂直于平面的z方向施加0.027Δc=0.007 ?.試算得到的能帶結構如圖10 所示,圖10(a)—(d)分別表示考慮泊松比前后,拉伸形變為2%的純黑磷烯、黑磷烯吸附Si 原子,電場作用的純黑磷烯、電場作用的黑磷烯吸附Si 原子的能帶結構.從圖10 可知,泊松比對于黑磷烯體系的能帶結構影響較小,其曲線形狀幾乎沒有改變,帶隙也僅具有極其微小的改變.這表明泊松比對黑磷的電子特性影響較小.如果未特殊說明,本文計算結果均未考慮泊松比.
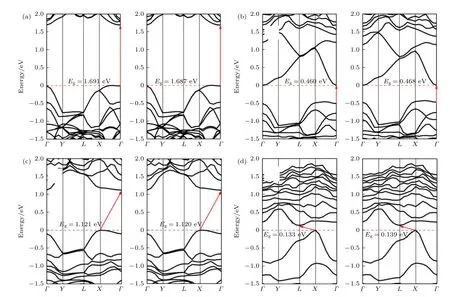
圖10 考慮泊松比前后、拉伸形變量為2%的黑磷烯能帶結構 (a)純黑磷烯;(b)黑磷烯吸附Si 原子;(c)電場與形變共作用的純黑磷烯;(d) 電場與形變共作用的黑磷烯吸附Si 原子Fig.10.Band structure of black phosphorene with 2% tensile deformation before and after considering Poisson’s ratio (a) Pure BP,(b) Si absorbed on BP,(c) pure BP with co-action of electric field and deformation,(d) Si absorbed on BP with co-action of electric field and deformation.
根據(1)式,首先計算了未吸附Si 的純黑磷烯體系在不同拉伸程度下的單原子結合能;根據(2)式,計算得到了拉伸黑磷烯吸附Si 原子吸附能;具體結果見表4.

表4 拉伸形變作用下純黑磷烯單原子結合能和黑磷烯吸附Si 原子吸附能Table 4.Monoatomic binding energy of black phosphorene and adsorption energy of Si adsorbed on black phosphorene under tensile deformation.
從表4 可以看出,在當前研究范圍內,對于純黑磷烯來說,其他體系的結合能的絕對值呈現出隨形變量增大而略微減小的趨勢,但變化較為微小,說明形變對于黑磷烯體系能量影響較小.類似地,對于黑磷烯吸附Si 原子體系來說,形變略微降低了吸附能力,但所有模型均屬于化學吸附,吸附過程存在電子轉移,伴隨化學鍵的生成,體系內原子重新排列.
3.3.2 黑磷烯吸附Si 原子拉伸體系電子特性
不同拉伸程度的純黑磷烯能帶如圖11(a)—(e)所示.拉伸改變了黑磷烯的準直接帶隙.當形變量增加為6%時,黑磷烯開始顯現出間接帶隙.形變量為10%的體系的CBM 能帶較為疏散,說明光產生電子的有效質量低,遷移率高,光生電子躍遷到CBM 的概率更大.除了8%的體系較為特殊,其余體系的帶隙大體呈現出隨形變量的增加先增加后略微降低的趨勢.而形變量為8%時,帶隙驟降.這是由于在拉伸程度為8%時,黑磷烯平面發生較大的幾何變形,其結構俯視圖如圖12 所示.這種波紋狀的幾何形變加劇了黑磷烯P 原子間電荷轉移,使其電子結構發生改變.觀察能帶結構還可發現,拉伸系統的價帶能量幾乎保持不變,導帶距電子勢壘的遠離或者接近導致了帶隙的減小或增大.這說明此時帶隙的改變主要依賴于應變引起的導帶底部能量變化.

圖11(a)—(e) 拉伸形變量為2%—10%的黑磷烯能帶結構;(f)—(j)拉伸形變量為2%—10%的黑磷烯吸附Si 原子體系能帶結構Fig.11.(a)-(e) Band structure of black phosphorene with 2%-10% tensile deformation;(f)-(j) band structure of Si adsorbed on black phosphorene with 2%-10% tensile deformation.
圖11(f)—(j)為黑磷烯吸附Si 原子拉伸體系能帶結構,可以看出變形較小時,帶隙打開較小,形變量為2%時帶隙為0.460 eV,且直接帶隙出現在價帶內.當形變量增加至4%時,帶隙驟增至1.750 eV,但此時VBM 和CBM 分別移動到點X和L處,對應直接帶隙.之后帶隙隨形變量增大而增加,但增幅較緩.形變量為8%和10%的體系的VBM 出現在Γ點,電子躍遷的所需動量增加.在當前研究范圍,純黑磷烯帶隙的范圍在1.578—1.771 eV,而黑磷烯吸附Si 原子帶隙在0—1.924 eV范圍變化,遠寬于純黑磷烯的變化范圍.這意味著吸附Si 原子的黑磷烯體系的間隙調諧比純黑磷烯的更有效.觀察形變量為8%的吸附體系的能帶結構發現,盡管幾何結構也出現了波浪狀的形變,但帶隙并未出現驟減的情況.這說明Si 原子的吸附體系電子結構穩定性較強,容易實現帶隙的穩定調控.
圖13 為純黑磷烯和黑磷烯吸附體系在拉伸應變作用下的態密度結構.對比純黑磷烯的PDOS圖,發現只有形變量為8%的結構在能量為—7 和—12 eV 的價帶內有局域內小波峰的減少與增加.其余體系DOS 形狀大體相同,僅在費米能級處為零的區域范圍有所不同,顯現出的帶隙大小與能帶圖相對應.觀察黑磷烯吸附Si 原子體系的DOS 發現,除了形變量為2%,其余體系在費米能級附近的—0.5—0 eV 處,Si 原子p 軌道的PDOS 與P 原子s 軌道的PDOS 重合,說明兩者軌道能量相近且發生雜化,對應著吸附后體系內存在共價鍵的特征.這也對應著帶隙在形變量為4%時的帶隙驟增.

圖12(a) 形變為8%的黑磷烯結構俯視圖;(b)形變為8%的黑磷烯電荷差分密度Fig.12.(a) Top view of the structure of 8% tensile deformation black phosphorene;(b) differential charge density of 8% tensile deformation phosphorene.

圖13(a)—(e) 拉伸形變量為2%—10%的純黑磷烯(BP)以及黑磷烯吸附Si 原子體系(BP-Si)態密度結構;(f)黑磷烯帶隙變化曲線Fig.13.(a)-(e) The DOS of black phosphorene (BP) and Si adsorbed on black phosphorene (BP-Si) with 2%-10% tensile deformation;(f) band gap curves of black phosphorene.
3.4 拉伸及電場共作用對黑磷烯吸附Si 原子體系影響
3.4.1 外加電場對黑磷烯吸附Si 原子體系影響
圖14 為電場作用下純黑磷烯與黑磷烯吸附Si 原子體系的能帶與態密度結構.在能帶圖中,藍色虛線代表未加電場的黑磷烯體系.從圖14 可以看出,電場使本征黑磷烯帶隙減小,且由直接帶隙轉變為間接帶隙.對于黑磷烯吸附Si 原子體系來說,電場作用沒有改變其零帶隙,但導帶中接近費米能級附近的能級變得稀疏.DOS 結構可以看出,Si 原子的DOS 貢獻與P 原子的DOS 曲線較為離散,未見強烈的雜化作用.
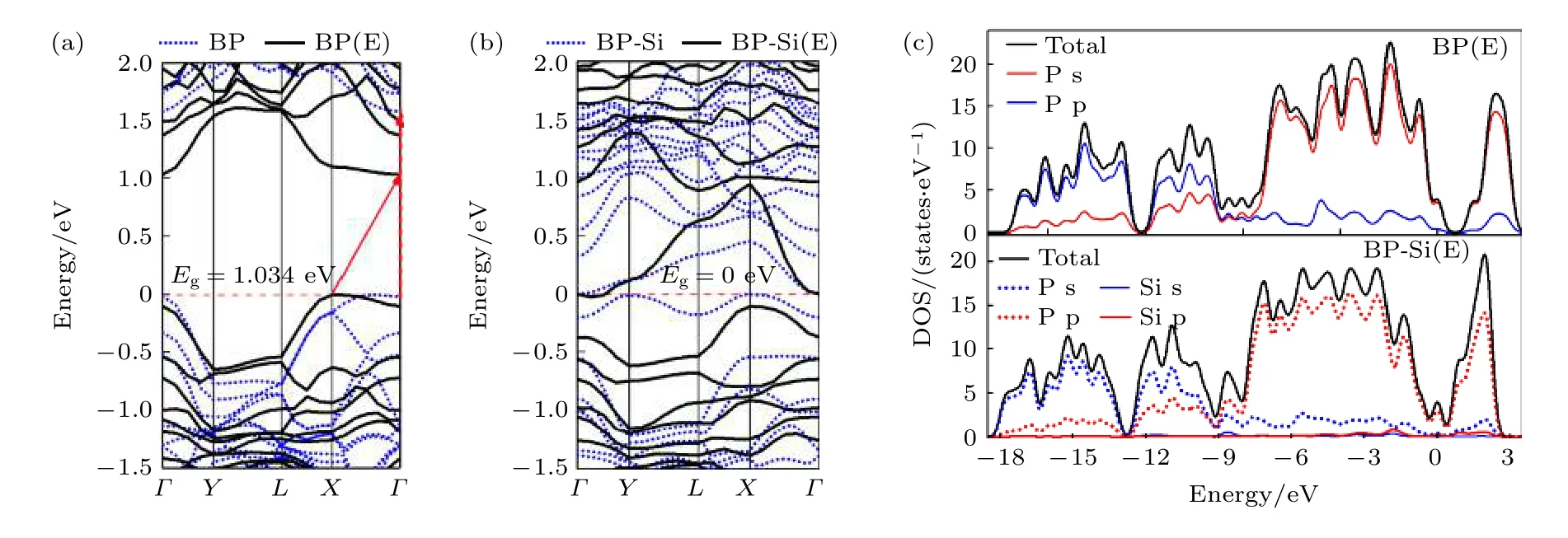
圖14 電場作用下純黑磷烯與黑磷烯吸附Si 原子體系 (a),(b)能帶結構,其中藍色虛線代表黑磷及黑磷烯吸附體系的能帶結構,黑色實現代表電場作用下黑磷烯及其吸附體系的能帶結構;(c) DOS 結構Fig.14.Si adsorbed on black phosphorene system and pure black phosphorene under electric field:(a),(b) Band structure,the blue dotted line represents the energy band structure of black phosphorus and black phosphorene adsorption system,and the black realization represents the energy band structure of black phosphorus and its adsorption system under the action of electric field;(c) DOS.
3.4.2 拉伸及電場共作用對黑磷烯吸附Si 原子體系影響
根據(1)式和(2)式,計算了純黑磷在不同拉伸量和外加電場共作用下的單原子結合能和黑磷烯吸附體系在不同拉伸量和外加電場共作用下黑磷烯的吸附能,具體結果列于表5.
在當前研究范圍內,電場的作用對黑磷烯及其吸附體系影響較大.比較表4 和表5 可得,外加電場使純黑磷烯體系的結合能大幅降低,相比于未加電場體系大約降低了3 eV.對有電場作用的純黑磷烯來說,體系結合能的絕對值隨拉伸變形量增加而減小,但變化較為微小.說明拉伸對電場作用的黑磷烯體系能量影響較小.外加電場對黑磷烯吸附Si 原子體系的吸附穩定性影響較小,吸附能變化不大.電場作用的吸附Si 原子黑磷烯體系的吸附能隨形變量的增強而減小.

表5 電場與拉伸共作用下純黑磷烯單原子結合能和黑磷烯吸附Si 原子吸附能Table 5.Single atom binding energy of black phosphorene and adsorption energy of Si adsorbed on black phosphorene system under the action of electric field and tensile.
圖15(a)—(e)顯示了純黑磷烯在拉伸應變和電場共同作用下的能帶結構.從圖15(a)—(e)可以看出,引入電場后所有體系變為間接帶隙.與未加電場的拉伸體系相比,電場的作用使體系帶隙大約減少了0.5 eV.圖15(f)—(j)為拉伸應變和電場共同作用下吸附體系的能帶結構,帶隙與未加電場的體系相比減少量在0.3—0.7 eV 不等.形變量增加至4%之后,帶隙幾乎無太大改變.說明此時形變量對于體系帶結構影響微弱.值得一提的是,在電場作用下形變量為10%的吸附體系由間接帶隙轉變為直接帶隙,VBM 與CBM 均出現在X點.引入電場后,黑磷烯體系的帶隙隨形變量的變化趨勢與未加電場的相一致,只是整體數值有所減小.說明電場會使體系的帶隙整體變窄.形變與電場共作用的黑磷烯體系態密度如圖16 所示.同未加電場的一樣,Si 原子的p 軌道與P 原子的s 軌道有劇烈的雜化,其成鍵具有共價性.
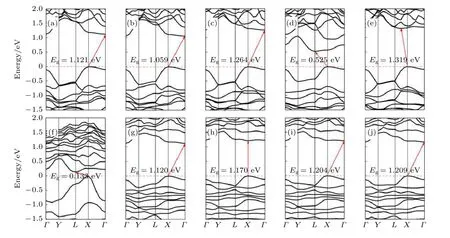
圖15(a)—(e) 電場作用下拉伸形變量為2%—10%的黑磷烯能帶結構;(f)—(j)電場作用下拉伸形變量為2%—10%的黑磷烯吸附Si 原子體系能帶結構Fig.15.(a)-(e) Band structure of black phosphorene with 2%-10% tensile deformation under electric field;(f)-(j) band structure of Si adsorbed on black phosphorene system with 2%-10% tensile deformation under electric field.
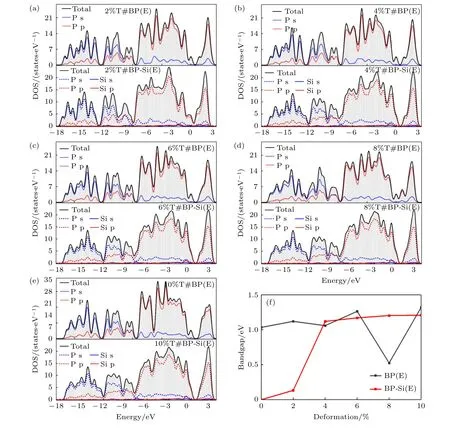
圖16(a)—(e) 電場作用下拉伸形變量為2%—10%的黑磷烯吸附Si 原子體系能帶結構;(f)黑磷烯帶隙變化曲線Fig.16.(a)-(e) Band structure of Si adsorbed on black phosphorene system with 2%-10% tensile deformation under the action of electric field;(f) band gap curves of black phosphorene.
4 結論
利用第一性原理方法,在結構優化的基礎上計算了不同吸附位黑磷烯吸附Si 原子體系的吸附能、鍵長及吸附高度.計算結果表明:Si 原子吸附在H 位吸附能最大,吸附最為穩定.對H 位吸附的黑磷烯吸附Si 原子模型進行能帶和態密度的計算表明,Si 原子的吸附使黑磷烯帶隙消失,實現了其由半導體向準金屬的轉變.這是由于吸附Si 原子后,幾何結構的改變破壞了黑磷烯的原始對稱性,軌道重疊程度的改變使P—P 鍵再雜化.Si 原子與黑磷烯間具有強烈的電子轉移,也加劇了黑磷烯中P 原子間的電荷轉移,改變部分原子成鍵類型.接著試圖通過形變及電場等手段調控黑磷烯吸附Si 原子體系的帶隙.為了更好地分析吸附體系電子特性,同時給出了本征黑磷烯的能帶結構與態密度.計算結果表明:拉伸形變和電場作用都會降低黑磷烯吸附體系的穩定性,也會顯著地改變其帶隙.在拉伸形變作用下,黑磷烯吸附Si 原子的帶隙被打開,再次完成由準金屬向半導體的轉變.其帶隙值隨形變量的增加而增加,在形變量為4%,Si 原子的p 軌道與P 原子的s 軌道有劇烈的雜化,帶隙增加幅度較大.之后隨形變量增加帶隙趨于穩定,增幅變小.吸附Si 原子的黑磷烯體系的間隙調諧比純黑磷烯的更有效,且其體系電子結構穩定性較強,容易實現帶隙的穩定調控.電場與拉伸共作用的情況下,電場的引入使黑磷烯體系帶隙變窄且均變為間接帶隙.當前研究范圍內,電場對于帶隙隨形變量的改變趨勢影響微弱.吸附Si 原子的黑磷烯體系帶隙可調性高于未吸附體系,且易于實現帶隙的穩定調控.
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中華詩詞(2019年7期)2019-11-25 01:43:04
中國外匯(2019年17期)2019-11-16 09:31:14
模具制造(2019年3期)2019-06-06 02:10:54
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
現代企業(2015年9期)2015-02-28 18:56:50
現代企業(2015年1期)2015-02-28 18:43:18
新高考·高一物理(2014年1期)2014-09-18 01:26:07
土木建筑工程信息技術(2013年2期)2013-10-17 03:14:12
