HPLC 法測定依帕司他片清潔驗證中殘留物
2021-11-17 02:50:20尤銳薛雯蔚馮小雪揚子江藥業集團南京海陵藥業有限公司江蘇南京210049
化工管理 2021年31期
尤銳,薛雯蔚,馮小雪(揚子江藥業集團南京海陵藥業有限公司,江蘇 南京 210049)
0 引言
糖尿病是一種常見的慢性非傳染性疾病,已成為嚴重的世界性公共衛生問題。《國務院關于實施健康中國行動的意見》[1]指出,我國是糖尿病患病率增長最快的國家之一。據國際糖尿病聯盟(IDF) 數據顯示,截至2019 年,在20 歲到79 歲的人群中,中國糖尿病患者數總人數約為1.164億[2]。然而,糖尿病致死原因主要歸于其并發癥。據世界衛生組織統計,糖尿病并發癥高達100 多種,是目前已知并發癥最多的一種疾病。糖尿病死亡者有一半以上是心腦血管所致,10% 是腎病變所致。因糖尿病截肢的患者是非糖尿病的10~20 倍。臨床數據顯示,糖尿病發病后10 年左右,將有30%~40% 的患者至少會發生一種并發癥,且并發癥一旦產生,藥物治療很難逆轉,因此強調盡早預防糖尿病并發癥。糖尿病周圍神經病變(diabetic peripheral neuropathy, DPN) 屬于糖尿病微血管并發癥,發病機制較為復雜,是老年2 型糖尿病患者反復住院的重要危險因素[3]。依帕司他片主要用于治療糖尿病周圍神經病變,是預防糖尿病并發癥的常用藥物。臨床研究證明[4],依帕司他聯合甲鈷胺治療糖尿病周圍神經病變效果顯著,可改善神經傳導速度與臨床表現,且患者無明顯不良反應,用藥安全性較高。
最大限度地降低藥品生產過程中污染、交叉污染是藥品生產質量管理的核心內容之一[5]。出于生產成本、生產空間的局限性,做到降本增效,在藥品生產過程中,不同藥品共用生產線和生產設備的情況不可避免。制定有效、便捷的清潔程序是防止污染、交叉污染的重要舉措之一。清潔驗證[6]是證明清潔程序能有效清潔設備并滿足其預定用途的直接證據。基于此,本文參照《日本藥典》中依帕司他含量測定的檢測方法,優化實驗條件,建立高效液相色譜法測定依帕司他片清潔驗證中的化學殘留。
1 實驗部分
1.1 主要儀器與試劑
Agilent 1260 高效液相色譜儀(安捷倫科技有限公司);XPE205DR 分析天平(梅特勒-托利多)。
磷酸二氫鉀(分析純,國藥集團化學試劑有限公司);磷酸氫二鈉(分析純,國藥集團化學試劑有限公司);乙腈(色譜純,TEDIA);乙醇(色譜純,TEDIA);N, N-二甲基甲酰胺(色譜純,TEDIA);布簽(TEXWIPE)。
1.2 實驗方法
1.2.1 色譜條件色譜柱:十八烷基硅烷鍵合硅膠為填充劑(Waters Xbrige C18 150mm×4.6 mm,5 μm);流動相:磷酸鹽緩沖液[0.05 mol/L磷酸二氫鉀溶液,用0.05 mol/L 磷酸氫二鈉溶液調節pH 值至6.5(0.05 mol/L 磷酸二氫鉀溶液與0.05 mol/L 磷酸氫二鈉溶液的體積比約為200∶85)]-乙腈(65∶35);檢測波長:294 nm;柱溫:25 ℃;進樣盤溫度:4 ℃;進樣量:50 μL;流速:1.0 mL/min;運行時間:10 min;洗針方式:沖洗端口,15 s;洗針溶液:乙腈。
1.2.2 標準溶液的配制和標準曲線的繪制
精密稱取依帕司他對照品約30.5 mg,置50 mL 容量瓶中,加N, N-二甲基甲酰胺適量,充分振搖使溶解,并稀釋至刻度,搖勻;精密量取上述溶液2 mL,置100 mL 容量瓶中,加流動相稀釋至刻度,搖勻,即得對照品貯備液。精密量取對照品貯備液5 mL 置50 mL 容量瓶中,加流動相稀釋至刻度,搖勻,即得對照品溶液。
精密量取依帕司他對照品貯備液適量,使制成濃度范圍在定量限至300% 濃度水平(約為3.66 μg/mL) 之間的6 個濃度水平的溶液。按1.2.1 項下色譜條件記錄色譜圖,以濃度(c) 為橫坐標,峰面積(A) 為縱坐標,計算回歸方程及相關系數。濃度級別及試驗結果如表1 所示。
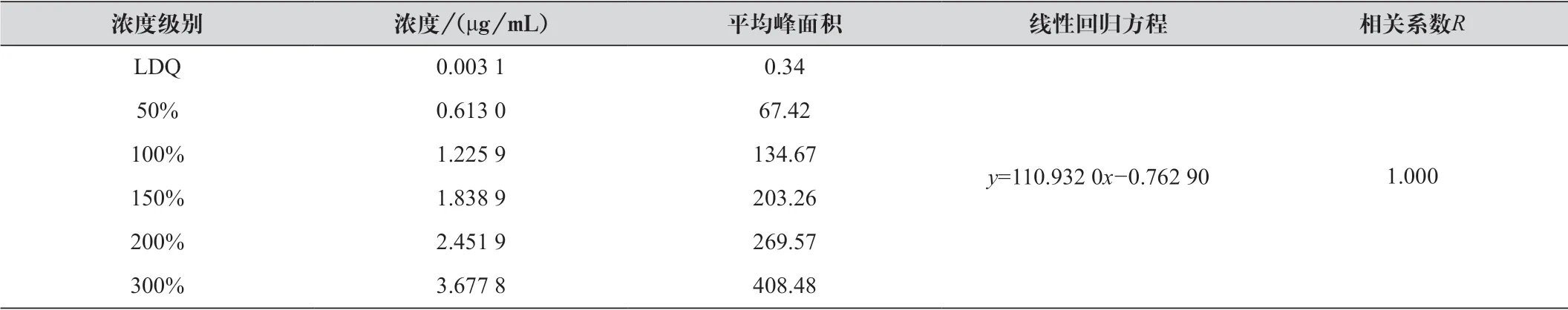
表1 不同濃度級別下依帕司他對照品溶液的線性關系
1.2.3 取樣回收率供試品溶液的制備
根據與產品直接接觸的設備、容器、工器具的材質,選擇316L 不銹鋼、聚四氟乙烯、有機玻璃和硅膠四種材質進行取樣回收率測試。
316L 不銹鋼板:精密量取對照品貯備液1 mL 均勻地滴加在316L 不銹鋼板的方塊區域上,方塊為10 cm×10 cm,常溫晾置約20 min,取布簽,用乙醇約0.2 mL 潤濕,將其按在涂好并晾置后的不銹鋼板上,盡量使布簽與取樣點表面完全接觸,平穩而緩慢地按圖1(a) 所示方向均勻擦拭一遍,然后翻轉布簽,用另一面按圖1(b) 所示方向進行第二次擦拭。同法重復3 次。取樣必須盡快完成以免溶劑揮發和污染物沉積到樣品表面。將在同一塊平板上取樣的布簽置于同一頂空瓶中,加入10 mL 流動相,超聲5 min,搖勻,過濾,作為取樣回收率供試品溶液。
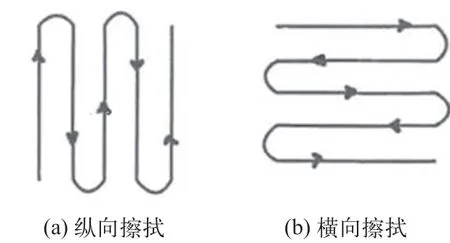
圖1 布簽擦拭示意圖
聚四氟乙烯板、有機玻璃板、硅膠板的相關供試品溶液制備過程同316L 不銹鋼板。
2 方法和結果
2.1 色譜條件的選擇和優化
以《日本藥典》[7]中依帕司他含量測定的檢測方法為參考,根據紫外分光光度計檢測結果,對檢測波長進行調整,確定以294 nm 作為檢測波長。在波長294 nm 下,考察了不同流動相比例對出峰時間和峰型的影響,發現原定流動相比例條件下布簽空白溶液在依帕司他主成分峰位置出現干擾峰。后調整流動相為磷酸鹽緩沖液-乙腈(65∶35),此時無明顯雜峰干擾,且出峰時間有所提高。在檢測波長和流動相比例同時優化的條件下,將主成分峰出峰時間提高近一倍,大大縮短了檢驗周期。
2.2 專屬性
取適量乙醇作為空白溶劑溶液。取布簽,用乙醇約0.2 mL 潤濕,將其分別按在試驗用的空白316L 不銹鋼板(10 cm×10 cm)、聚四氟乙烯板(10 cm×10 cm)、有機玻璃板(10 cm×10 cm)、硅膠板(10 cm×10 cm) 上,按1.2.3 所述方法制備,即為布簽空白溶液。
取空白溶劑溶液、布簽空白溶液、對照品溶液適量,用1.2.1項下色譜條件進行色譜分析,記錄各色譜圖。結果表明,空白溶劑、空白布簽及空白材質對依帕司他殘留物含量的測定均不產生干擾,供試品溶液中主峰與相鄰峰的分離度為2.45,分離情況良好。
2.3 溶液穩定性
取4℃下放置的對照品溶液和供試品溶液,分別于0、6、12、18、24、30、36、42、48 h分別進樣采集,記錄色譜圖,結果顯示,4 ℃下放置的對照品溶液及供試品溶液在48 h 內穩定,其各時間點的保留時間與初始測得值的比值在0.98~1.02 之間,各個時間點測得的主成分峰面積與初始峰面積的相對平均偏差均小于1.0%。
2.4 檢測限與定量限
以信噪比約為3∶1 時的濃度作為該方法的檢測限,以信噪比不小于10 時的濃度作為該方法的定量限。結果顯示,該方法的檢測限濃度為0.000 766 2 μg/mL,限度為0.000 076 62 μg/cm2;定量限濃度為0.003 065 μg/mL,限度為0.000 306 5 μg/cm2。
2.5 重復性
取依帕司他對照品溶液適量,按1.2.1 項下色譜條件進行色譜分析,平行進樣6 次,記錄其色譜圖中主成分的峰面積,計算出其RSD。結果表明,6 次測得主成分峰面積RSD 為0.1%,精密度較好,符合測定的要求。
2.6 取樣回收率
按1.2.3 項下配制取樣回收率供試品溶液,各材質分別由兩名人員各擦拭3 次,得到每種材質6 份供試品溶液。以外標法進行定量分析,結果如表2 所示。結果表明,取樣回收率均大于70%,RSD 值小于20%,符合制劑生產中清潔到位的要求。
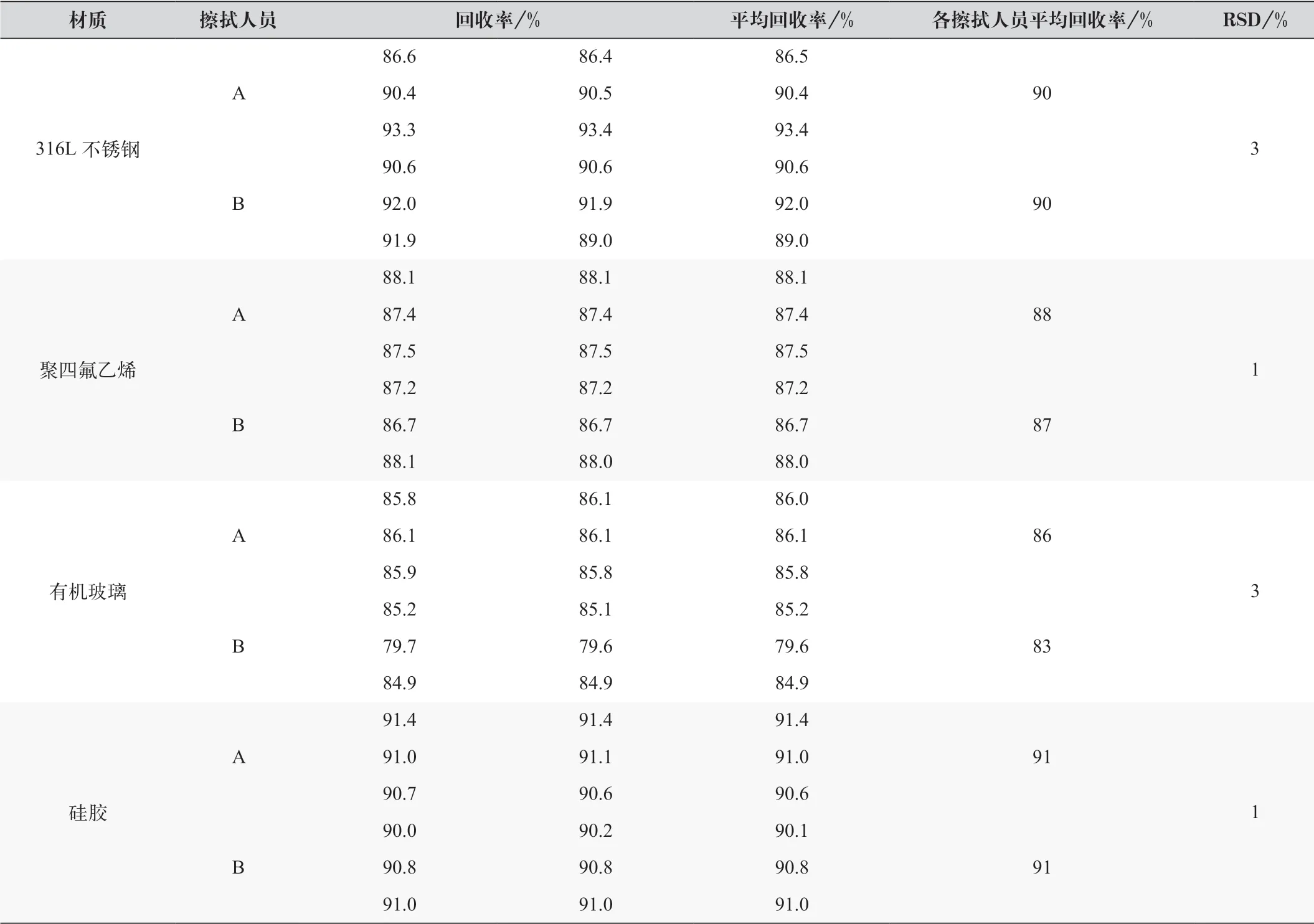
表2 各材質取樣回收率結果
3 結語
建立了高效液相色譜法測定制劑生產清潔過程中依帕司他片的主成分殘留量,通過優化色譜條件,大大縮短了檢測周期和時間成本。對該方法專屬性、線性、穩定性、檢測限與定量限、精密度和回收率進行驗證,結果表明該方法滿足《藥品生產驗證指南》[8]相關要求,適用于依帕司他片清潔驗證的殘留物檢測,為大生產過程中依帕司他片清潔驗證提供了一種準確、快速且可靠的殘留物檢測方法。
猜你喜歡
中老年保健(2022年5期)2022-08-24 02:35:42
中老年保健(2022年1期)2022-08-17 06:14:56
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中老年保健(2021年5期)2021-08-24 07:07:20
中老年保健(2021年9期)2021-08-24 03:51:04
中老年保健(2021年7期)2021-08-22 07:42:16
中老年保健(2021年11期)2021-08-22 03:15:16
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
