六方氮化硼納米片改進(jìn)QuEChERS法凈化草莓中10種農(nóng)藥殘留
2021-11-10 01:25:54趙燕芳李慧娟謝含儀陳相峰
趙燕芳,李慧娟,謝含儀,陳相峰
(齊魯工業(yè)大學(xué)(山東省科學(xué)院)山東省分析測(cè)試中心,山東濟(jì)南 250014)
草莓是一種小型水果,具有吸引人的顏色和獨(dú)特的風(fēng)味,富含有益于人體健康的化合物,并且熱量值相對(duì)較低,因而成為最受人們歡迎的水果之一[1]。草莓可以以新鮮、冷凍或加工等形式存儲(chǔ)、食用,因?yàn)楹性S多重要的膳食成分,包括礦物質(zhì)、纖維,以及豐富的植物素(鞣花酸、花青素、槲皮素和兒茶素)和維生素(抗壞血酸和葉酸),所以被列為多酚和抗氧化能力的膳食來(lái)源之一[2]。為了提高草莓產(chǎn)量和質(zhì)量,農(nóng)藥在草莓生長(zhǎng)過(guò)程中起著不可替代的作用;然而,不規(guī)范使用農(nóng)藥會(huì)導(dǎo)致草莓農(nóng)藥殘留甚至超標(biāo),嚴(yán)重影響環(huán)境安全、出口貿(mào)易以及消費(fèi)者身體健康[3-4],因此,草莓中農(nóng)藥殘留的檢測(cè)是一個(gè)非常重要的課題。
常用于農(nóng)藥殘留檢測(cè)的技術(shù)包括氣相色譜法[5]、液相色譜法[6]、氣相色譜質(zhì)譜法[7]和液相色譜質(zhì)譜法[8]。樣品前處理是農(nóng)藥殘留檢測(cè)的關(guān)鍵環(huán)節(jié)。為了避免復(fù)雜基質(zhì)的干擾,樣品前處理的關(guān)鍵步驟是樣品提取液的凈化。由于植物樣品中含有大量的色素、氨基酸、生物堿等成分,因此在提取過(guò)程中這些物質(zhì)會(huì)隨著目標(biāo)有害物共提取出來(lái)[9]。該類共提取雜質(zhì)會(huì)給檢測(cè)過(guò)程帶來(lái)諸多問(wèn)題,如污染色譜柱、離子源以及干擾目標(biāo)物的檢測(cè)結(jié)果,造成假陽(yáng)性等問(wèn)題。最常用的提取技術(shù)有固相萃取(SPE)法[10]、液-液萃取法[11]、滲透凝膠色譜[12]和QuEChERS(快速-簡(jiǎn)單-廉價(jià)-高效-耐用-安全)法[13]等。傳統(tǒng)的固相萃取法的選擇性不強(qiáng),無(wú)法建立綜合的多殘留分析方法。液-液萃取法基于相似相溶原理,不適于性質(zhì)差異較大的多種殘留的凈化。常規(guī)的凝膠滲透色譜儀器昂貴,且耗時(shí)長(zhǎng),消耗有機(jī)試劑。使用固相萃取法和離線滲透凝膠色譜法時(shí),存在工作效率低和操作繁瑣等問(wèn)題,因此QuEChERS法以操作簡(jiǎn)便和快速的特點(diǎn),成為最常用的前處理方法之一。QuEChERS法通常采用N-丙基乙二胺(PSA)、十八烷基硅烷鍵合硅膠(C18)、石墨化碳黑等作為固相吸附提取液中的雜質(zhì),在去除色素及中弱極性干擾物時(shí)存在凈化效果不足問(wèn)題,因此需要開(kāi)發(fā)具有吸附多種雜質(zhì)新型固定相,實(shí)現(xiàn)提取液的凈化。
六方氮化硼(hexagonal boron nitride, h-BN)納米材料以等數(shù)目的氮原子和硼原子通過(guò)sp2雜化方式結(jié)合成六元環(huán),每個(gè)B、N原子提供一個(gè)p軌道形成共軛π鍵[14],具有良好的生物相容性和吸附性能。Jia等[15]制備了大比表面積(大于1 400 m2/g)的多孔h-BN納米材料,用于吸附水中的多種有機(jī)物,發(fā)現(xiàn)該材料能吸附其自身質(zhì)量33倍的污染物。Wang等[16]將h-BN作為基質(zhì)用于基質(zhì)輔助激光解析離子化質(zhì)譜法(MALDI-TOF)分析小分子,較傳統(tǒng)基質(zhì)具有較小的背景干擾,提高了靈敏度。Furuhashi等[17]發(fā)現(xiàn)h-BN保留的磷酸化肽段的數(shù)量為C18和石墨化碳黑之和,可用來(lái)吸附化學(xué)性質(zhì)較寬范圍的肽段。h-BN納米材料的獨(dú)特性質(zhì)已經(jīng)引起了國(guó)內(nèi)外學(xué)者的廣泛關(guān)注,但是尚未見(jiàn)其用于樣品基質(zhì)凈化的應(yīng)用。
本文中首次利用一種大比表面積的二維h-BN納米片作為吸附固定相,采用分散固相萃取技術(shù)改進(jìn)QuEChERS法,建立一種草莓樣品中多種農(nóng)藥殘留的快速、簡(jiǎn)便、有效前處理方法,進(jìn)一步結(jié)合高效液相色譜串聯(lián)質(zhì)譜法開(kāi)發(fā)10種農(nóng)藥殘留的分析方法。
1 實(shí)驗(yàn)
1.1 儀器與試劑
主要儀器包括:高效液相色譜串聯(lián)質(zhì)譜儀,Triple QuadTM 5500 AB SCIEX型,美國(guó)AB SCIEX有限公司;管式爐,OTF-1200X型,安徽合肥科晶材料科技有限公司;烘箱,DGX-9053B型,上海南榮實(shí)驗(yàn)設(shè)備有限公司;恒溫水浴鍋,6HZ-A型,上海博迅實(shí)業(yè)有限公司醫(yī)療設(shè)備廠;離心機(jī),MULTIFUGE XIR型,美國(guó)Thermo Scientific公司;真空干燥箱,DZF-6094型,上海一恒科學(xué)儀器有限公司。
主要試劑包括:甲醇(CH3OH,CAS 67-56-1)、乙腈(CH3CN,CAS 75-05-8),色譜純,美國(guó)新天地有限公司;氯化鈉(NaCl,CAS 7647-14-5),優(yōu)級(jí)純,國(guó)藥集團(tuán)化學(xué)試劑有限公司;無(wú)水硫酸鈉(Na2SO4,CAS 7487-88-9),分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司;有機(jī)污染物相關(guān)化學(xué)品標(biāo)準(zhǔn)物質(zhì),純度大于95%(質(zhì)量分?jǐn)?shù)),德國(guó)Dr公司。單標(biāo)標(biāo)準(zhǔn)溶液的儲(chǔ)備液質(zhì)量濃度為10 mg/L。
1.2 儀器條件
色譜條件:色譜柱,Thermo Fisher Scientific C18型, 粒徑為2.6 μm, 2.1 mm×100 mm(內(nèi)徑×長(zhǎng)度); 流動(dòng)相為甲酸銨(濃度為10 mmol/L)和乙腈,洗脫模式采用線性梯度洗脫。洗脫條件見(jiàn)表1,洗脫速率為0.3 mL/min,進(jìn)樣體積為5 μL,色譜柱溫度為30 ℃,樣品盤溫度為10 ℃。

表1 高效液相色譜洗脫條件
質(zhì)譜條件: 離子源為電噴霧(ESI)離子源,使用正離子掃描模式。電噴霧電壓為5 500 V,離子源溫度為500 ℃。氣簾氣壓力為138 kPa,碰撞氣壓力為50 kPa,霧化氣壓力為345 kPa,輔助氣壓力為345 kPa。入口電壓為10 V,碰撞室出口電壓為13 V。監(jiān)測(cè)模式為多反應(yīng)監(jiān)測(cè)(MRM)。
1.3 h-BN納米片的制備
h-BN納米片制備方法依據(jù)文獻(xiàn)[18]并在其基礎(chǔ)上進(jìn)行改進(jìn),步驟如下:在容積為50 mL的離心管中加入甲醇(10 mL)和水(10 mL)的混合液,然后分別加入物質(zhì)的量分別為0.15、0.05 mol的尿素、硼酸,上述混合液在室溫下水浴振蕩1 h,然后在50 ℃烘箱中加熱12 h,將得到的白色固體轉(zhuǎn)移到石英舟中在管式爐中煅燒,氣氛為氮?dú)猓褵郎囟葹?00 ℃,保溫時(shí)間為2 h,得到的白色粉末用無(wú)水乙醇離心洗滌3次,在溫度為70 ℃時(shí)真空干燥2 h。
1.4 樣品前處理
1)草莓樣品處理。將草莓樣品洗干凈,取可食用部分置于攪碎機(jī)里攪碎,勻漿,裝入潔凈的離心管內(nèi),密封并標(biāo)明標(biāo)記。將試樣于-20 ℃冷凍保存。
2)標(biāo)準(zhǔn)溶液配制。稱取10 g勻漿后的空白草莓漿,置于50 mL離心管中,加入一定量的農(nóng)藥殘留標(biāo)準(zhǔn)溶液,渦旋1 min,加入10 mL乙腈,渦旋1 min,再加入2 g氯化鈉和4 g無(wú)水硫酸鎂,離心分離,取上清液置于50 mL離心管中,加入4 mg h-BN納米片,渦旋1 min,在轉(zhuǎn)速為8 000 r/min時(shí)離心分離,上清液過(guò)孔徑為0.22 μm的濾膜后上機(jī)檢測(cè)。
3)加標(biāo)回收率實(shí)驗(yàn)。稱取10 g勻漿后的空白基質(zhì)樣品,置于50 mL離心管中,加入一定量的農(nóng)藥殘留標(biāo)準(zhǔn)溶液,渦旋1 min,加入10 mL乙腈,渦旋1 min,再加入2 g氯化鈉和4 g無(wú)水硫酸鎂,離心分離,取上清液置于50 mL離心管中,加入4 mg h-BN納米片,渦旋1 min,在轉(zhuǎn)速8 000 r/min下離心分離,上清液過(guò)孔徑為0.22 μm的濾膜后上機(jī)檢測(cè)。
2 結(jié)果與討論
2.1 h-BN納米片的表征
h-BN納米片的微觀形貌圖像及吸附-脫附等溫曲線見(jiàn)圖1。由圖1(a)、(b)可以看出,h-BN納米片具有二維的類似石墨烯的形貌; 由圖1(c)可以推算出h-BN納米片的比表面積很大,數(shù)值約為2 000 m2/g。h-BN納米片這種類石墨烯的結(jié)構(gòu)及大的比表面積有助于對(duì)檢測(cè)樣品中雜質(zhì)的吸附和對(duì)復(fù)雜基質(zhì)凈化。

(a)掃描電子顯微鏡圖像

(b)透射電子顯微鏡圖像
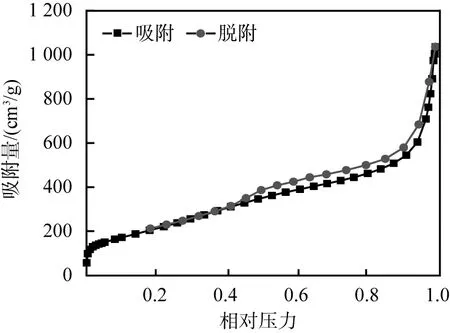
(c)吸附-脫附等溫曲線圖1 六方氮化硼納米片的微觀形貌圖像及 吸附-脫附等溫曲線
2.2 儀器條件的優(yōu)化
2.2.1 色譜條件的優(yōu)化
在液相色譜分析中,流動(dòng)相的組成是一個(gè)重要因素,不僅影響目標(biāo)物的色譜峰型和分離效果,也會(huì)影響檢測(cè)器的信號(hào)響應(yīng)和檢測(cè)靈敏度。色譜優(yōu)化過(guò)程以信號(hào)響應(yīng)強(qiáng)度、峰型和干擾程度作為評(píng)價(jià)效果的依據(jù)。選用濃度為10 mmol/L的甲酸銨和乙腈作為流動(dòng)相梯度洗脫,得到草莓樣品中10種農(nóng)藥殘留化合物的保留時(shí)間色譜圖,如圖2所示。

圖2 草莓樣品中10種農(nóng)藥殘留的保留時(shí)間色譜圖
2.2.2 質(zhì)譜條件優(yōu)化
優(yōu)化三重四極桿質(zhì)譜是一項(xiàng)艱巨的任務(wù),原因是每個(gè)目標(biāo)化合物都需要特定的實(shí)驗(yàn)條件進(jìn)行分析。為了找出保留時(shí)間和分析物峰之間的最佳分辨率,使用混合標(biāo)準(zhǔn)溶液在全掃描模式下進(jìn)行初步實(shí)驗(yàn)。串聯(lián)質(zhì)譜探測(cè)器具有較高的選擇性和靈敏度,重點(diǎn)考慮的是母離子、子離子的選擇,以及最佳反應(yīng)的碰撞能量的優(yōu)化。質(zhì)譜參數(shù)選擇首先對(duì)化合物進(jìn)行全面掃描,然后選擇每個(gè)分析物的母離子,繼而優(yōu)化碰撞能量電壓來(lái)生成產(chǎn)物離子。進(jìn)一步優(yōu)化每個(gè)化合物在MRM獲取過(guò)程中的特征離子躍遷和碰撞能量,進(jìn)行定量分析。各化合物參考保留時(shí)間、監(jiān)測(cè)離子對(duì)(同一化合物的母離子對(duì)應(yīng)不同的子離子)、去簇電壓(DP)、碰撞能量電壓(CE)參考值見(jiàn)表2。
2.3 凈化條件優(yōu)化
2.3.1 h-BN納米片用量
為了得到較大的回收率及可以重現(xiàn)的色譜峰強(qiáng)度,去除基質(zhì)中的干擾物質(zhì)是必要的。本文中提出用QuEChERS法凈化草莓樣品中基質(zhì)干擾物,無(wú)水硫酸鎂用于去除基質(zhì)中的水分,h-BN納米片用于去除其他干擾物質(zhì),如脂肪酸、色素和其他基質(zhì)化合物。研究發(fā)現(xiàn),h-BN納米片可以作為QuEChERS法中的替代材料,用于分散固相萃取基質(zhì)中干擾物來(lái)凈化草莓樣品。圖3所示為h-BN納米片用量條件優(yōu)化結(jié)果。從圖中可以看出:少量的h-BN納米片 (小于4 mg)不能獲得最大回收率,原因可能是吸附劑的用量較少,不能很好地凈化樣品基質(zhì);當(dāng)h-BN納米片用量為4 mg時(shí),回收率達(dá)到最大值,并且經(jīng)過(guò)處理的最終樣品顏色透明;當(dāng)h-BN納米片用量過(guò)多時(shí),可能導(dǎo)致吸附大多數(shù)加標(biāo)的農(nóng)藥。本文中選用4 mg為h-BN納米片的最佳用量。
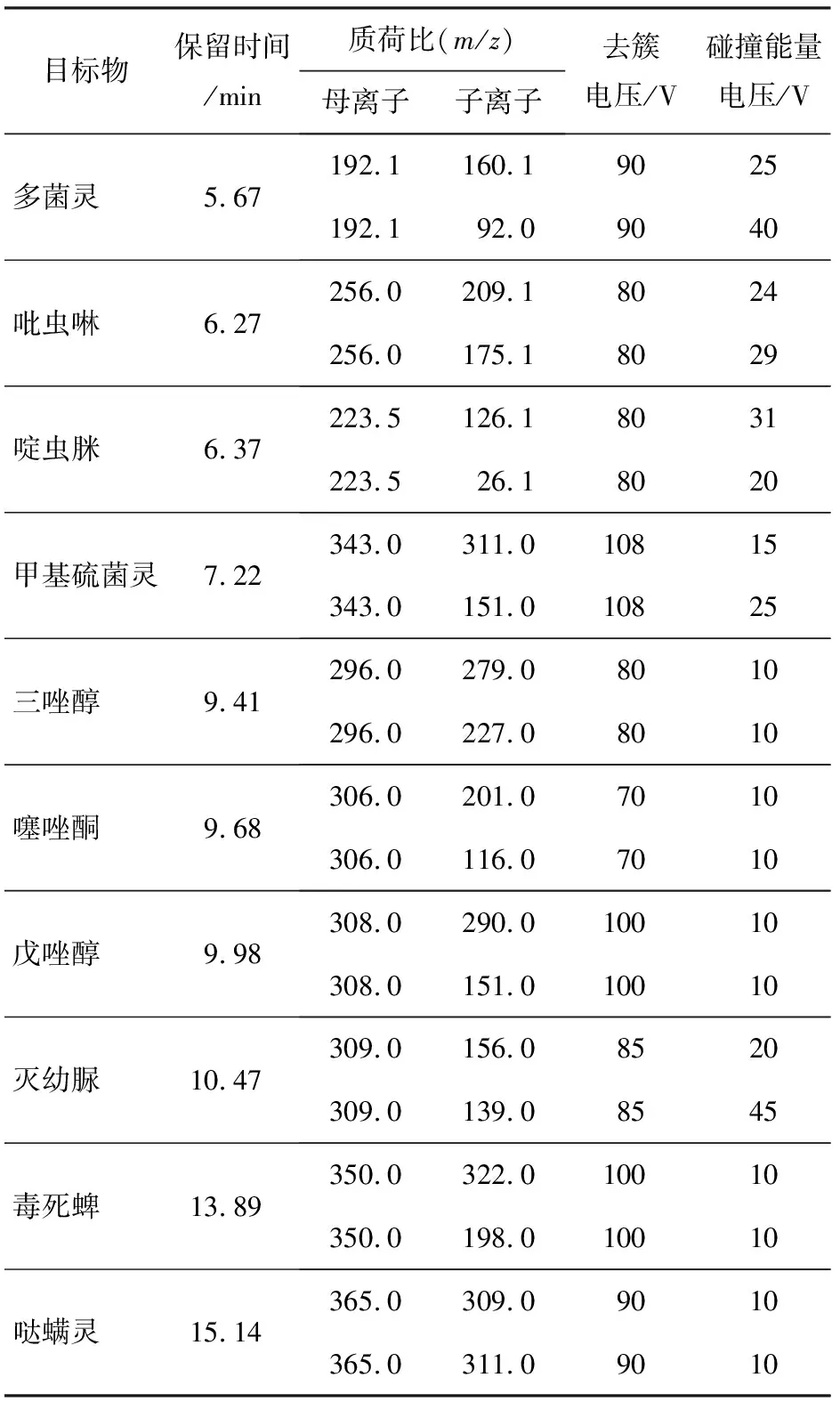
表2 草莓樣品中10種農(nóng)藥殘留的保留時(shí)間、監(jiān)測(cè)離子對(duì)、去簇電壓、碰撞電壓參數(shù)
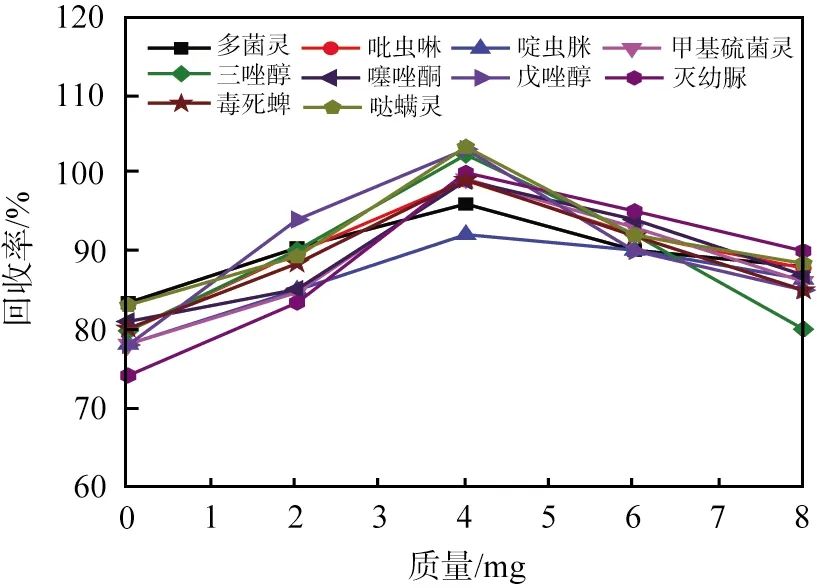
圖3 10種農(nóng)藥殘留回收率與六方氮化硼納米片用量的關(guān)系
2.3.2 凈化時(shí)間
凈化時(shí)間也是影響凈化效果的一個(gè)重要因素。凈化時(shí)間為20~100 s時(shí)對(duì)不同目標(biāo)物(質(zhì)量比為100 mg/kg)回收率的影響見(jiàn)圖4。由圖可以看出:凈化初始時(shí)目標(biāo)物的回收率較小,原因是凈化時(shí)間過(guò)短,不能使h-BN納米片充分凈化草莓樣品基質(zhì),干擾了目標(biāo)物的測(cè)定。凈化時(shí)間為60 s時(shí),凈化劑充分發(fā)揮了凈化作用,目標(biāo)物回收率最大。隨著凈化時(shí)間繼續(xù)增加,目標(biāo)物的回收率減小,原因可能是凈化時(shí)間過(guò)長(zhǎng),h-BN納米片對(duì)目標(biāo)物有部分吸附。實(shí)驗(yàn)最終選定60 s作為最佳凈化時(shí)間。
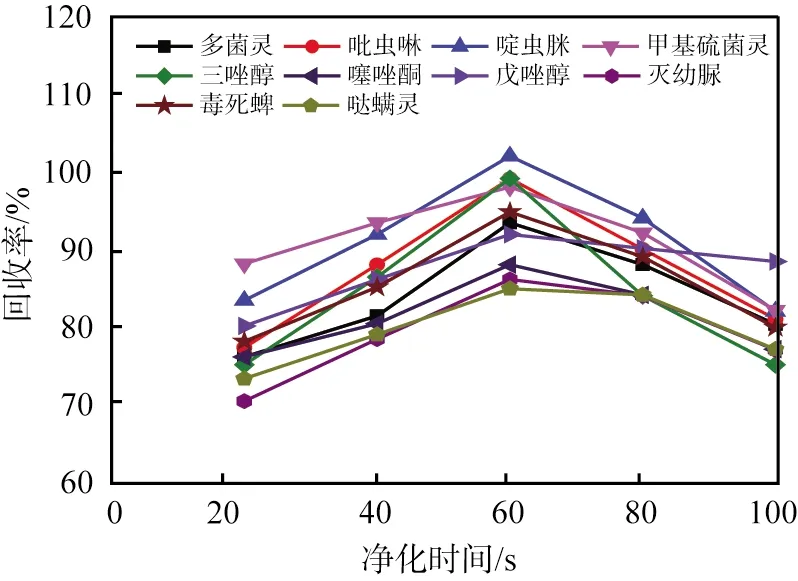
圖4 10種農(nóng)藥殘留回收率與凈化時(shí)間的關(guān)系
2.4 方法建立
草莓中10種農(nóng)藥殘留的分析結(jié)果見(jiàn)表3。從表中可以看出,在最優(yōu)的實(shí)驗(yàn)條件下,10種農(nóng)殘質(zhì)量比線性范圍均為0.1~500 mg/kg,相關(guān)系數(shù)r為0.999 2~0.999 8,方法檢出限(信噪比為3)為0.000 5~0.01 mg/kg,定量限(信噪比為10)為0.002~0.003 mg/kg。采用質(zhì)量比為100 mg/kg的10種農(nóng)藥殘留進(jìn)行重復(fù)性實(shí)驗(yàn),日內(nèi)相對(duì)標(biāo)準(zhǔn)偏差為2.56%~5.98%(n=5,n為測(cè)定次數(shù)),日間相對(duì)標(biāo)準(zhǔn)偏差為2.24%~5.32%(n=5),不同批次合成的h-BN納米片的重復(fù)性相對(duì)標(biāo)準(zhǔn)偏差為2.25%~6.25%(n=5)。上述結(jié)果表明,h-BN納米片可以改進(jìn)QuEChERS法凈化草莓樣品中農(nóng)藥殘留污染物。
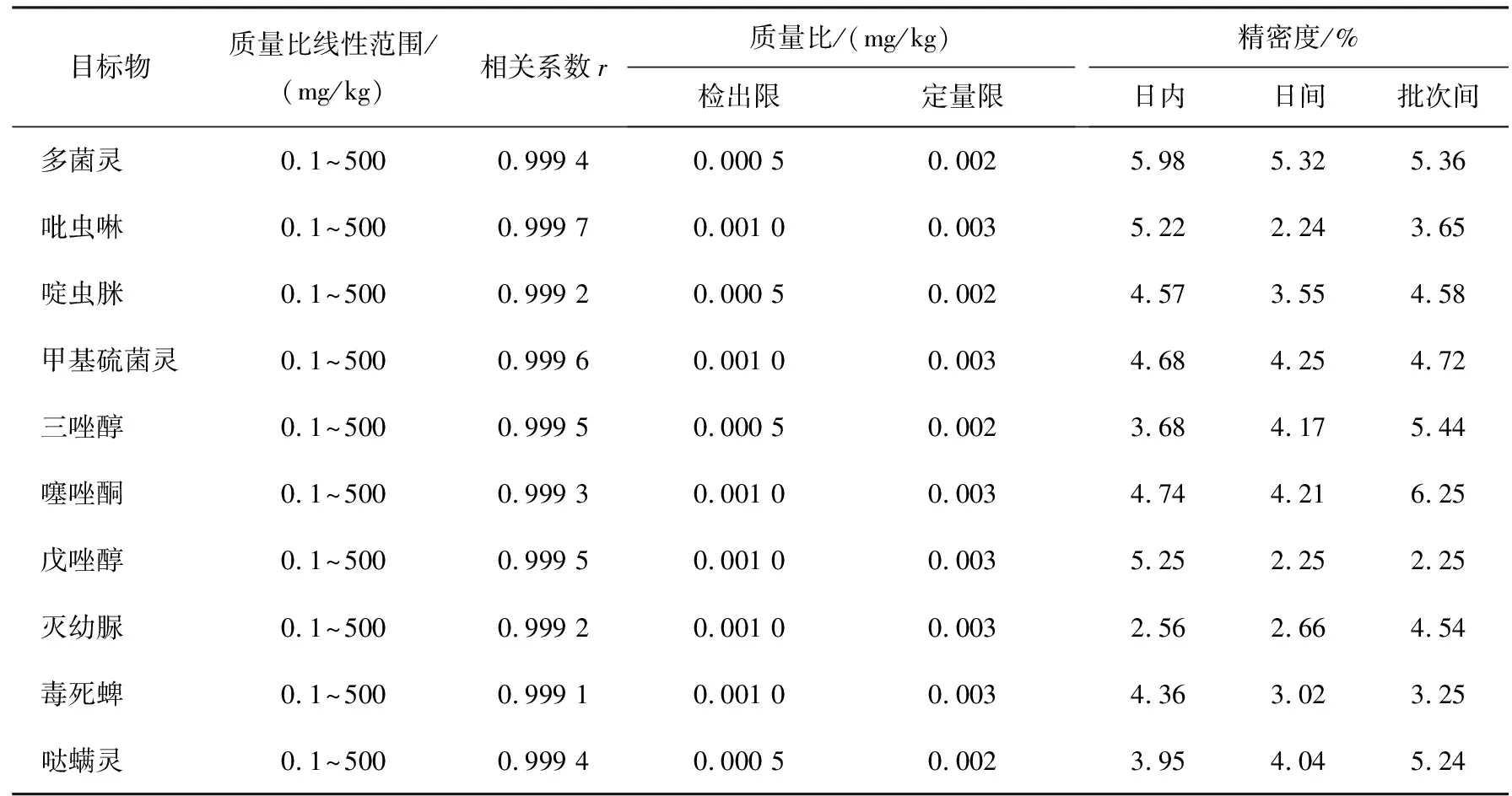
表3 草莓中10種農(nóng)藥殘留的分析結(jié)果
2.5 樣品分析
將基于h-BN納米片改進(jìn)的QuEChERS法用于草莓樣品中10種農(nóng)藥殘留污染物的凈化,在草莓樣品中加入農(nóng)藥殘留標(biāo)準(zhǔn)物質(zhì)的量作為加標(biāo)量(質(zhì)量比分別為1、10、50 mg/kg),對(duì)于加標(biāo)樣品也進(jìn)行同樣的前處理。進(jìn)一步結(jié)合高效液相色譜串聯(lián)質(zhì)譜儀分析實(shí)際樣品中痕量農(nóng)藥殘留污染物,結(jié)果見(jiàn)表4。由表可以看出,樣品的回收率為86.03%~106.3%,表明該方法結(jié)果穩(wěn)定、可靠,可應(yīng)用于草莓實(shí)際樣品中痕量農(nóng)藥殘留的分析。
3 結(jié)論
本文中首次利用h-BN納米片改進(jìn)的QuEChERS法,建立了一種快速、靈敏的草莓樣品中多種農(nóng)藥殘留分析方法,得到以下主要結(jié)論:
1)h-BN納米片用于分散固相萃取法,可以有效凈化基質(zhì)中農(nóng)藥提取物。萃取后的凈化過(guò)程主要是去除基質(zhì)中的干擾物質(zhì),而不是提取和分離分析物。
2)h-BN納米片可以快速、有效地吸附樣品中色素等干擾物,凈化基質(zhì),并結(jié)合高效液相色譜串聯(lián)質(zhì)譜法可以分析樣品中痕量農(nóng)藥殘留。
3)該方法的線性范圍、精密度、回收率均符合農(nóng)藥分析的要求,結(jié)果證實(shí)了該方法的可行性,可用于草莓產(chǎn)品中農(nóng)藥殘留的常規(guī)檢測(cè)和監(jiān)測(cè),為痕量農(nóng)藥分析和樣品清理找到有效途徑,有望廣泛應(yīng)用于各種水果中微量農(nóng)藥的監(jiān)測(cè)。
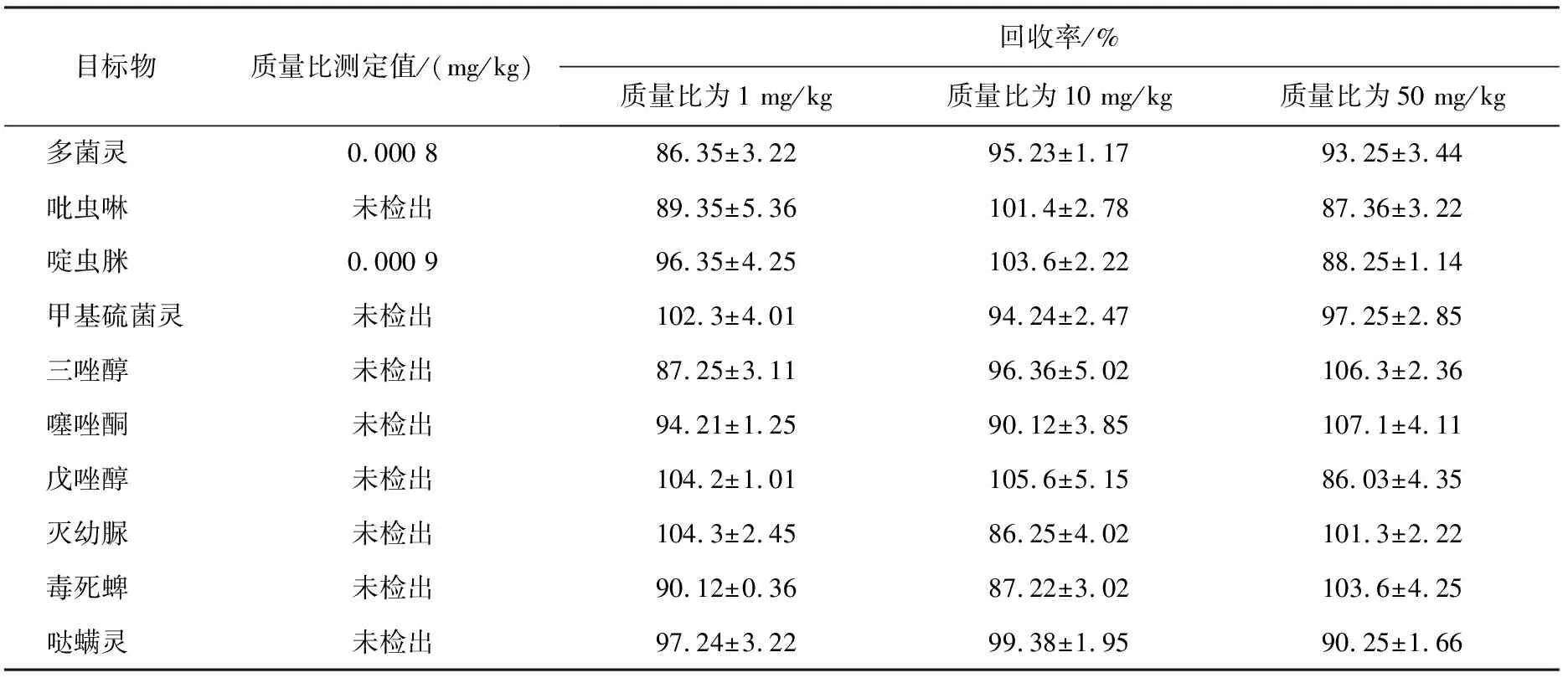
表4 草莓樣品中10種農(nóng)藥殘留污染物不同加標(biāo)量(質(zhì)量比)回收率結(jié)果
