鹵醇脫鹵酶的研究進展
2021-11-05 13:56:08王龍興賈紅華
生物加工過程 2021年5期
王龍興,賈紅華,韋 萍
(南京工業大學 生物與制藥工程學院,江蘇 南京 211800)

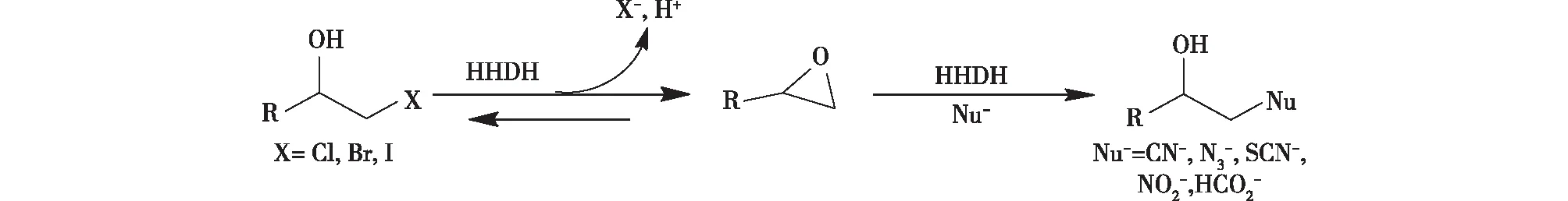
圖1 HHDHs催化的脫鹵與開環反應
在早期的研究中,利用產HHDH的微生物來降解環境中含有鹵代有機化合物的污染物,使其在環境治理方面具有重要應用價值。之后,通過對HheC突變、結構解析以及動力學等方面的研究,揭示了HHDH的結構特點以及催化脫鹵、開環的機制[8-9],發現HHDHs在催化制備手性環氧化物和光學純的β-取代醇方面具有極高的應用價值[10],尤其是在制備降脂藥阿托他汀的前體、手性環氧氯丙烷等方面,這使得HHDHs應用前景更廣闊。因此,本文綜述了近年來關于HHDHs在來源分布、結構與機制、分子改造以及生物催化應用等方面的研究進展,為相關的研究者提供參考。
1 HHDHs的來源與分布
HHDHs從發現到酶家族的擴展,經過了幾十年的發展,最初僅發現了HheA、HheA2、HheB、HheB2和HheC這5種代表性的HHDHs。它們分別來自Corynebacteriumsp. N-1074[11]、Arthrobactersp. AD2[8]、Arthrobactersp. AD2[11]、Arthrobactersp. AD[8]、AgrobacteriumradiobacterAD1[8]和Rhizobiumsp. NHG3[12]。這5種酶又被分A、B和C三個亞類,亞類內的酶序列相似度超過97%,亞類間的相似性卻低于33%,但與SDR家族其他酶具有較高的相似性。
近年來,隨著各類數據庫中基因序列的爆發式增長,通過生物信息學手段挖掘新酶已成為主要途徑[13],因而HHDHs酶家族的數量迅速擴增。目前文獻中已報道的HHDHs達到77種之多[14-17],其中已有20多種酶得到詳細的表征,不同酶的底物譜及立體選擇性都有差異(表1),大多數HHDHs脫溴的活力高于脫氯的活力,這是由于C—Cl的鍵能高于C—Br,使得C—Cl不易斷裂;另外,有研究表明HHDHs不能催化C—F斷裂而脫氟[18]。

表1 已表征HHDHs的底物譜及活性
根據進化分析,HHDHs酶家族已由A類擴增到G類,共7個亞類,各亞類之間的HHDHs相似性為25%~45%。HHDHs家族的擴增主要集中在A、B、D和E亞類,雖然目前HheC是目前研究最多的亞類,但迄今仍然僅一種(圖2)。目前發現的HHDHs中,來源于海洋宏基因組的HHDHs集中分布于B類和E類,而其他亞類的HHDHs則多來源于土壤、海洋、污水或淡水淤泥等環境的細菌,其中以變形菌門居多。從HHDHs的來源來看,多為鹵素富集區域,這恰與微生物能夠在含有有毒的鹵代有機物中利用HHDHs代謝鹵代醇生長繁殖的習性相適應,暫時還未在其他生物中發現HHDHs的蹤跡。

圖2 HHDH酶家族進化分析
Koopmeiners等[19]在對17種新酶進行表征的時候發現,酶的立體選擇性似乎和酶的分類存在著關聯,在對2-氯-苯乙醇進行脫鹵反應時,B類、E類新酶的立體選擇性為S型,而A類和D類的新酶卻是R型。但是Xue等[14-15]對挖掘的新酶進行表征的時候發現,即使同亞類的酶針對同種底物也有不同的立體選擇性。就立體選擇性而言,HheC仍是催化脫鹵反應的對映選擇性最高(E>100)的酶[15, 19]。但HheC的熱穩定和催化反應的最適溫度明顯低于HheA3、HheA5、HheD、HheD3和HheD5[19],這也是HheC進行分子改造時的重點研究領域。從新酶的表征數據來看,還未發現HHDHs的分類和功能之間的規律。
2 HHDHs的結構與催化機制
HHDHs的活性中心是由Ser-Tyr-Arg構成的催化三聯體,這個結構與SDR家族非常相似,不同的是SDR家族是Ser-Tyr-Lys[7,9]。通過對已報道的HHDHs的氨基酸序列進行多序列比對發現,Tyr與Arg之間總被3個氨基酸隔開[13],而SDR家族中,超過86%酶的Tyr與Lys之間也相隔3個氨基酸[6-7];在HHDHs中,Ser與Tyr之間相隔12個氨基酸,但SDR家族中Ser與Tyr的相對位置卻不是那么保守[13]。因而由催化三聯體結構組成的S-X12-Y-X3-R基序被認為是辨別HHDHs與SDR家族的重要特征之一(圖3)。此外,HHDHs在N端還擁有1個含芳香族氨基酸(Phe或Tyr)的寬敞的陰離子結合口袋,這一結構替代了SDR中富含Ala或Gly的NAD(P)輔酶結合位點的位置,成為另一個辨別HHDHs的特征序列T-X4-(F/Y)-X-G[9](圖3)。陰離子結合口袋通過主鏈上的酰胺鍵形成的氫鍵、芳香族氨基酸殘基帶的正電荷以及水分子的結合來穩定陰離子。雖然在酶的結構中有許多氫鍵供體,但是芳香族的氨基酸殘基能使碳鹵鍵更輕易地斷裂[23]。

圖3 HHDHs的特征基序
迄今為止,人們已測定了HheA[24]、HheA2[25]、HheB[24]、HheC[9]和HheG[26]的晶體結構,由此發現,HHDHs是由一對二聚體組成的一個同源四聚體(圖4)。與SDR家族一樣,HHDHs擁有一個典型的羅斯曼折疊結構——7個或者8個α-螺旋包裹著6個或7個平行的β-折疊。雖然酶活性中心在酶的內部,但是底物能通過一個通道與活性中心的氨基酸殘基結合,通道中的氨基酸殘基則影響HHDHs的底物特異性和產物立體選擇性[27-29]。
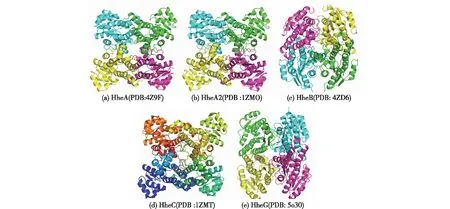
圖4 HHDHs的晶體結構


圖5 HHDHs的催化機制
3 HHDHs的分子改造
野生型酶的催化性能往往無法適應工業化應用的需求,隨著定向進化等技術的發展,通過對HHDHs進行酶分子改造以提升其性能是一種重要途徑。HheC在制備阿托他汀藥物前體方面具有很高的價值,但野生型酶的立體選擇性及酶活力較差。2007年,Fox等[31]首次采用具有革命性的技術——ProSAR驅動的定向進化技術,針對HheC催化(S)-4-氯-3羥基丁酸乙酯轉化為(R)-4-氰基-3-羥基丁酸乙酯的反應進行分子改造,在眾多的突變體中至少篩選到含有35個突變體,產物的體積生產率提高了4 000倍左右。在該研究的基礎上,Schallmey等[29]構建了含有37個突變位點的經典突變酶HheC-2360,通過酶學性質表征和結構解析研究發現,HheC-2360催化(S)-氯-3羥基丁酸乙酯脫鹵的kcat提高了3.1倍,在CN-介導下催化(S)-3,4-環氧丁酸酯開環的kcat提高了10倍,立體選擇性由HheC的R型轉變為HheC-2360的S型,并且熱穩定性提高了8 ℃。以HheC-2360的晶體結構為模型進行分子動力學模擬,與產物(3R,5R)-6-氰基-3,5-二羥基己酸叔丁酯進行分子對接,辨識出關鍵氨基酸殘基位點,然后對關鍵位點進行定點突變,篩選出突變體V84G/W86F,其催化活性比HheC-2360提高了15倍,Km值降低了4/5,kcat增加了3.3倍,使酶與底物的親和性大大提高并擴大了酶與底物的結合空間,產率提高了2倍[32]。此外,HheAAM(HheA12)通過迭代飽和突變后,突變體M4-HheAAM對(3R,5R)-6-氯-3,5-二羥基己酸叔丁酯的比酶活達到了0.89 U/mg,比野生型提高13倍[33]。
另外,提高HheC的穩定性也是近年來研究的重要內容。湯麗霞團隊的Wu等[34]采用組合定向進化的策略,先對野生型HheC進行易錯PCR的隨機突變,篩選出6株陽性突變體,共包含8個突變點,即F12P、D182E、Q87R、N157V、K52E、M252L、F136I和N159R;然后通過迭代飽和突變對這8個突變位點進行突變,發現隨著突變點增加,酶的熱穩定性隨之提高,最終篩選擁有8個突變位點的突變體ISM-4,在未影響其酶活力的前提下,較野生型的酶,其熱穩定性大大提高,65 ℃的半衰期提高了3 400倍,tm提高了18 ℃。依據在HheC的表面選取兩個相鄰的帶電荷且與活性中心距離不超過1.2 nm的原則,Wang等[35]選取K203和K204兩個位點同時突變為帶有電荷的R、D、E和K,從48個克隆篩選出CSL1 (K203R)、CSL2(K204R)、CSL3 (K203R/K204R)3個陽性突變體;與野生型相比,CSL2在55 ℃的半衰期提高了近6.9倍且不影響酶活性,CSL3在55 ℃的半衰期提高了3.4倍,但是其催化1,3-二氯-2-丙醇的kcat值提高了1.8倍;據動力學模擬分析,這是由于突變點的氨基酸殘基變化優化了蛋白質表面電荷間的相互作用和氫鍵作用,使整體結構更穩定,從而提高HheC的熱穩定性。與傳統的易錯PCR等定向進化技術相比,Arabnejad等[36]采用FRESCO方法,通過能量計算、二硫鍵預測以及分子動力學模擬對HheC的突變位點進行理性設計,不僅使突變后的HheC熱穩定性提高了23 ℃,而且耐有機溶劑;突變體HheC-H12在其最適溫度50 ℃下,其活性是野生型的2倍,在50%甲醇、50%乙腈、25%DMSO、25%二惡烷、25%四氫呋喃和25%二甲基甲酰胺溶液中能維持活性不變,其中在50%乙腈溶液中能保持5 h以上無活性損失,且動力學常數和底物范圍與野生型相近;HheC-H12包含了12個突變點:A29L、D39K、E64R、S68R、Q87R、A93T、C153N、A158V、E190T、E197K、V99I和V236I,其中C153N對穩定性的貢獻最大,它不僅改善了殘基間的相互作用力,還在溶劑間形成穩定的氫鍵,它也成為迄今是在穩定性方面表現最佳的突變體。
HheG是近年來通過基因挖掘發現的新酶,對氧化檸檬烯和環氧環己具有很高的活性,但是其穩定性不佳,限制了它的應用。Solarczek等[37]計算模擬分析HheG的晶體結構,篩選了20個氨基酸殘基位點進行飽和突變,最終發現T123位點的突變體T123G、T123F、T123Y、T123W和T123H的tm不僅提高了7~14 ℃,其酶活力還提高了3倍,其中T123G催化反應的初始速度更是提高了5倍。根據分子動力學模擬發現,靠近HheG活性位點裂縫位置有一個快速移動的環,處于將裂縫部分覆蓋或不覆蓋的狀態。當裂縫部分被覆蓋的時候,酶活性位點不易于底物接觸,而環路遷移率和位置直接受123位氨基酸殘基的影響[35]。
4 HHDHs的應用
4.1 HHDH制備阿托他汀藥物前體
HHDHs的一個重要應用是催化合成降血脂藥阿托他汀側鏈基團(R)-4-氰基-3-羥基丁酸乙酯。早期研究采用HheC及其突變體[31]和(S)-型酮還原酶聯合催化下合成了(R)-4-氰基-3-羥基丁酸乙酯。而采用HheA的全細胞催化 (S)-4-氯-3-羥基丁酸乙酯轉化為(R)-4-氰基-3-羥基丁酸乙酯的轉化率和產率分別可達95%和85%,但是細胞的消耗較大[38]。后來采用1,3-二氯-2-丙醇為原料,HheC先催化脫鹵并在CN-介導下開環合成(S)-4-氯-3-羥基丁腈,然后再次脫鹵開環合成(R)-4-氰基-3-羥基丁酸乙酯[39]。在填充柱中填充HHDH固定化酶,連續催化(S)-4-氯-3-羥基丁酸乙酯合成(R)-4-氰基-3-羥基丁酸乙酯,10 d后底物轉化率達90.6%,經過精餾提純可將收率達到98.2%、純度達到99.3%、光學純度達到99.1%[40]。另外,HheC突變體和腈水解酶AtNIT2共同級聯催化,可直接將(S)-4-氯-3-羥基丁酸乙酯轉化成(R)-3-羥基戊二酸,更有利于后續阿托伐他汀側鏈基團的合成[41]。因為(R)-4-氯-3-羥基丁酸乙酯作為中間體合成阿托伐他汀的路徑需要-78 ℃的極端條件,Luo等[32]便改進了路徑,利用HheC2360-V84G/W86F催化(3S,5R)-6-氯-3,5-二羥基已酸叔丁酯轉化為(3S,5R)-6-氰基-3,5-二羥基己酸叔丁酯,從而避免了原先路線后續的苛刻反應條件。
4.2 HHDH制備環氧氯丙烷

4.3 HHDH在生物催化其他方面的應用

5 總結和展望
HHDHs因其既能催化鄰鹵代醇脫鹵成環,又能催化環氧化物開環的特殊機制,使其成為生物催化領域一種重要的生物催化劑。它不僅可用于降解鹵代有機污染物,還可廣泛應用于降脂藥阿托伐他汀前體、環氧氯丙烷以及其他環氧化物、β-取代醇等化合物的制備。它的催化三聯體構成的基序S-X12-Y-X3-R以及N端的陰離子口袋構成的基序T-X4-(F/Y)-X-G是辨識HHDH序列的兩個重要特征,也是基于數據庫挖掘新酶篩選的標志。隨著基因測序技術的發展,生物信息數據庫中基因數據呈現爆炸式的增長,通過HHDHs序列特征在數據庫中挖掘新的HHDHs是當前的主要方向。可以預計未來會有更多的HHDHs家族其他成員被挖掘出來,不斷拓展HHDHs的來源。
從目前已表征的酶來看,野生型酶的穩定性和立體選擇性不佳,很難滿足工業生產的需求。通過ProSAR驅動的定向進化技術、易錯PCR、迭代飽和突變、FRESCO法等方法對酶分子改造,HHDH的穩定性和立體選擇性均有顯著提高。隨著蛋白質工程技術的快速發展,定向進化技術越來越成熟以及相應的高通量篩選設備來越先進,預計未來將改造出更多具有優異性能的HHDHs,以滿足工業過程的需要。另外,酶固定化技術也提升了HHDHs的催化性能,使HHDH可以回收,多次重復利用,在生物反應器中,可連續性操控生物催化反應,更加適應工業化大規模生產的需求。
由于脫鹵反應進行高通量篩選HHDHs的顯色試劑以及開環反應所需的親核試劑多為劇毒、易制爆藥品,普通實驗室硬件設施難以滿足條件,一定程度上阻礙了HHDHs的快速發展。開發安全便捷的分析方法也將是研究HHDHs的重要內容。
隨著酶工程技術越來越成熟以及高通量篩選等設備越來越先進,基于數據庫挖掘新酶,通過分子改造及固定化等技術獲取更多能夠滿足工業需求的HHDHs,完善HHDH生物催化的工藝條件將成為研究人員未來關注的焦點,預計未來將有許多可適應醫藥化工等行業大規模生產需求的HHDH商業酶產品。
