利用固體NMR研究一種萜類有機分子在聚乙烯中的吸附行為
2021-11-03 08:18:46宋建會劉宣伯姚雪容張韜毅張龍貴
石油化工 2021年10期
關鍵詞:結構
宋建會,劉宣伯,姚雪容,李 娟,張韜毅,張龍貴
(中國石化 北京化工研究院,北京 100013)
聚乙烯(PE)是一種應用廣泛的高分子材料,作為包裝材料具有價格便宜、力學性能優異、加工方式多樣、透明度和柔軟度好等特點,在農業、食品、藥品等領域具有廣泛的應用[1-2]。包裝材料的阻隔性能需滿足一定的要求,而氧氣、水蒸氣、揮發性化合物等小分子在膜中的吸附和遷移都會影響包裝產品的貨架壽命[3]。前期已有關于PE吸附醛、甲基酮、甲酯和含硫化合物等氣味分子的研究報道,影響吸附的因素包括分子尺寸、極性、溶解性、氣味分子的濃度以及聚合物的性質(如形貌、結晶度和極性等)[4-5]。本課題組曾報道了萜類有機分子在不同聚烯烴材質中的滲透行為,提出萜類有機分子的擴散系數和滲透系數與聚合物基體的結晶度和玻璃化轉變溫度有關[6]。目前對PE包裝材料吸附的研究主要關注在吸附量及吸附速率方面,而有機分子吸附對PE基體的結構影響以及擴散機理等尚缺乏深入認識。
固體NMR技術是研究固態高分子材料結構和分子動力學的一種非常重要和有效的手段[7-8],在PE的相結構研究方面具有獨特的優勢,可同時對PE結晶相和非晶相的結構及分子運動進行研究[9-11]。
本工作采用固體NMR技術考察了PE吸附一種萜類氣味有機分子前后的相結構及分子運動變化情況,以探索有機分子在PE中的吸附機理。
1 實驗部分
1.1 試劑
PE:密度0.924 g/cm3,熔體流動指數(10 min)1.9 g(190 ℃,2.16 kg),中海殼牌石油化工有限公司;C10H18O:萜類氣味有機分子,室溫下為固體,易揮發,閃點65 ℃,密度0.992 g/cm3,在25 ℃下的飽和蒸氣壓為3.12 Pa,云南林緣香料有限公司。
1.2 吸附實驗
采用Carver公司Auto4533型實驗室熱壓機和厚度為0.5 mm的壓片模具制備PE薄片。將壓好的薄片置于裝有C10H18O的密封容器中進行常溫吸附,至薄片恒重后用于NMR測試。吸附飽和后的PE中C10H18O的含量約為0.2%(w),吸附后的試樣記為PE/OM。試樣粉碎后裝入核磁轉子(氧化鋯,Kel-F帽子)中壓實,測試過程中由于密閉充實的環境,且轉子對小分子不吸附,可忽略吸附的C10H18O的脫附。
1.3 固體NMR測試
固體NMR測試在Agilent公司600 MHz Premium Compact+型核磁共振波譜儀上進行,1H和13C的共振頻率分別為599.89,150.86 MHz,室溫下測試。4 mm探頭,轉速8 kHz,1H和13C的90°脈寬均為4 μs。以金剛烷為標樣優化90°脈沖及Hartmann-Hahn條件,且13C的化學位移(δ)以金剛烷的次甲基信號(δ=38.56)為參考定標。
13C CP/MAS(交叉極化、魔角旋轉和高功率去偶)NMR實驗的接觸時間為1 ms,等待時間為4 s。13C SPE/MAS(單脈沖實驗,魔角旋轉和高功率去偶)NMR的循環等待時間為1 000 s。
13C自旋-晶格弛豫時間(T1)的測量采用Torchia脈沖序列,利用Origin的指數衰減公式對信號強度和間隔時間進行擬合得到PE各相的13CT1值。
2 結果與討論
2.1 C10H18O對PE基材結構的影響
PE,C10H18O及 PE/OM 的13C CP/MAS NMR譜圖見圖1。從圖1可看出,C10H18O的譜圖較復雜,因此未對信號進行詳細歸屬。對于PE,由于半晶聚合物的相組成按經典描述僅包含無定形相和結晶相,因此δ=32.9,31.1處的峰分別對應PE的正交晶相和非晶相[12]。PE/OM的譜圖中只有PE的信號,這是因為即使PE基材吸附C10H18O至飽和后,PE/OM中的C10H18O含量依然很低,僅約為0.2%(w),因此很難觀察到C10H18O的信號。吸附的C10H18O對PE的信號不造成明顯干擾,因此吸附前后試樣的NMR譜圖差異可直接反映PE結構的變化。
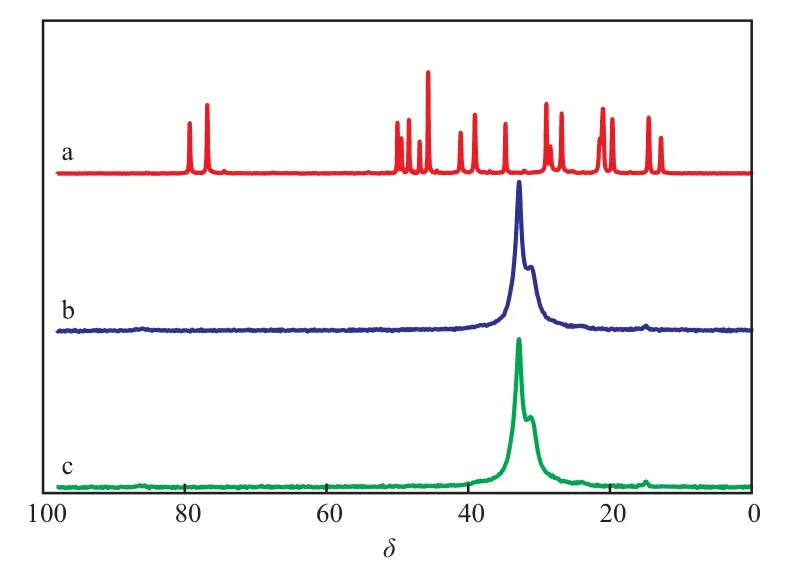
圖1 13C CP/MAS NMR譜圖Fig.1 13C CP/MAS NMR spectra.
由于13C CP/MAS NMR譜圖受交叉極化動力學的影響無法定量[13],可通過PE和PE/OM的13C SPE/MAS NMR譜圖(即去偶13C NMR譜)(見圖2)對結構進行定量研究,等待時間為1 000 s以確保碳核可以完全弛豫。
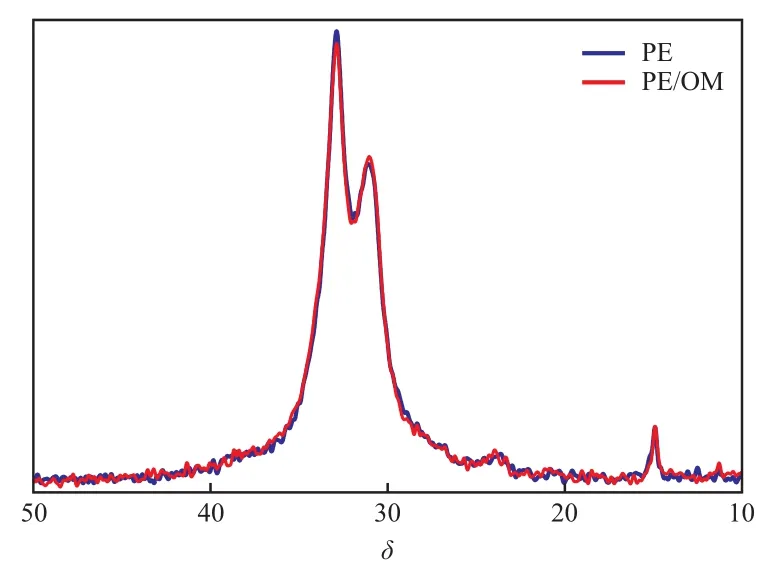
圖2 PE和PE/OM的13C SPE/MAS NMR譜圖Fig.2 13C SPE/MAS NMR spectra of PE and PE/OM.
從圖2可以看出,PE和PE/OM的13C SPE/MAS NMR譜圖存在一些差異,為了得到相區的含量需對譜圖進一步進行分峰處理。PE作為半晶高分子,它的相結構除了結晶區和無定形區,在二者之間還存在界面區,界面區的分子鏈排列具有一定的有序程度,而且同時具有晶區和無定形區的結構特征[14-15]。利用dmfit軟件對PE和PE/OM的13C SPE/MAS NMR譜進行分峰擬合,將譜圖分成晶區、界面區和無定形區,分峰結果見圖3。從圖3可看出,該結果與Mattozzi等[16]報道的PE相結構一致:δ=33.1處的峰歸屬為晶區,δ=31.7處的峰歸屬為界面區,δ=31.2處的峰歸屬為無定形區,各相區的含量見表1。
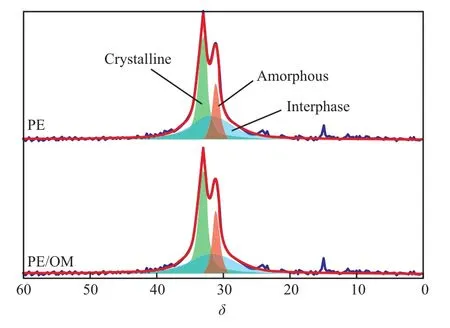
圖3 13C SPE/MAS NMR譜圖的分峰擬合Fig.3 Peak fitting of 13C SPE/MAS NMR spectra.
從表1可看出,相比于PE,PE/OM晶區的含量基本不變,界面區的含量略有減少,無定形區的含量有所增加,這是由于吸附的C10H18O使PE的界面區部分發生“溶解”,形成更加無序的無定形區。
2.2 C10H18O對PE基材分子運動的影響
固體NMR還常用于研究體系的微觀分子運動,是研究高分子體系中的分子運動非常有效的手段之一。通過測量13CT1研究PE基體在吸附C10H18O前后的分子運動變化。通常弛豫敏感的是分子運動的頻率而非模式,體系中某一種弛豫行為常對應多種分子運動過程[17]。T1與體系中的高頻運動相關,弛豫現象遵循Bloembergen-Purcell-Pound理論[18],對于PE,T1越大表明分子運動越慢。
圖4為不同衰減時間時PE的13C NMR譜圖。從圖4可看出,信號強度隨衰減時間的延長呈e指數函數衰減。晶區(δ=32.9)和無定形區(δ=31.1)信號強度的衰減趨勢存在差異,晶區信號強度的衰減速率比無定形區慢得多,這是由于晶區和無定形區的鏈段運動性存在差異:晶區鏈段運動受到晶格限制,表現出剛性,因此具有較長的13CT1,表現為信號強度衰減慢;無定形區的鏈段運動劇烈,表現出柔性,因此具有較短的13CT1,表現為信號強度衰減快。

圖4 Torchia法測量PE的13C T1Fig.4 13C NMR spectra of PE by Torchia pulse sequence.
對PE和PE/OM的13C T1結果用積分面積進行計算,積分范圍為δ=26~42,積分面積隨時間的衰減曲線見圖5。從圖5可看出,PE/OM衰減比PE快。PE的13CT1衰減曲線往往無法用單指數衰減公式擬合,因為實際數據包含不只一個組分,因此需要用多指數公式進行擬合[19]。在13C SPE/MAS NMR譜圖分峰結果中,PE的相結構分為晶區、界面區和非晶區,相應地會有3種不同弛豫行為的分子運動。因此,將衰減曲線分成3個組分擬合13CT1:
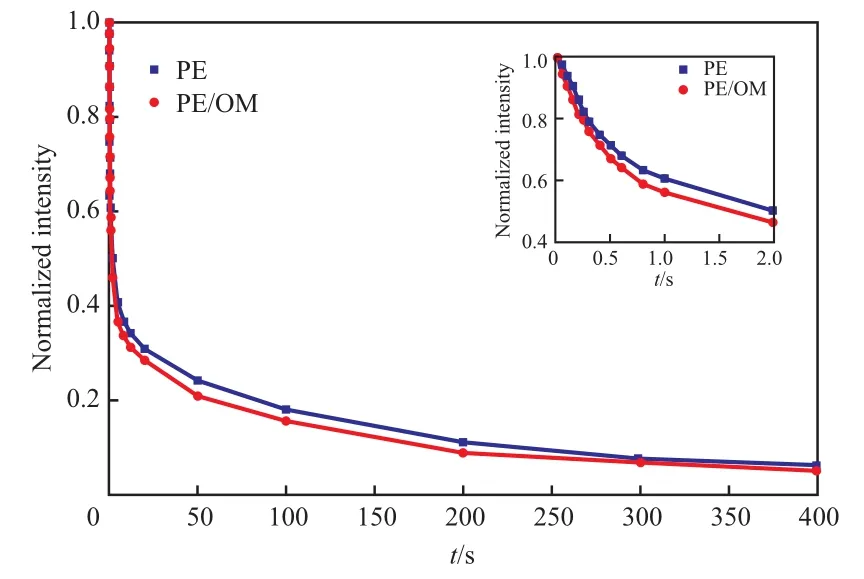
圖5 PE和PE/OM的13C T1衰減曲線Fig.5 13C T1 decay curve of PE and PE/OM.
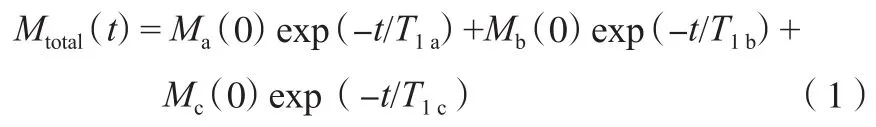
式中,t為延遲時間,s;Mtotal(t)為t時的總縱向磁化強度;Mi(0)(i=a,b,c)為各組分的初始磁化強度;T1i(i=a,b,c)為各組分的自旋-晶格弛豫時間,s。
PE和PE/OM的13CT1見表2。從表2可看出,由13CT1衰減曲線擬合得到的PE/OM三個組分的含量與PE相比變化不大,但三個組分的13CT1均比PE的短,這與圖5中PE/OM曲線衰減更快相符。由于PE的結晶分子受晶格限制,運動能力弱,因此最大的13CT1對應PE的晶區;無定形區的分子鏈運動最快,因此最小的13CT1對應PE的無定形區;界面區的鏈段運動能力處于晶區和無定形區中間,因此中間的13CT1對應PE的界面區。此處擬合出的三個組分的含量與SPE/MAS NMR譜圖分峰擬合的結果有所差異,這是因為兩種實驗方法及數據處理的原理不同:13C SPE/MAS NMR譜圖分峰是根據晶區、界面區和無定形區的化學環境(化學位移)不同;而13CT1擬合則是根據不同的相區結構對應不同的弛豫行為,由13CT1衰減曲線擬合得到擬合度最高的三個相區含量。在相區比例變化不大的情況下,由13CT1衰減曲線擬合得到的含量僅有參考意義。吸附C10H18O后PE基材的界面區和無定形區的13CT1均變小,這主要是由于吸附的C10H18O的“溶劑化”作用使界面區和非晶區的的分子鏈運動變快。而吸附后晶區的13CT1也變小,表明溶劑還是一定程度地進入了晶區,使晶區的分子運動能力加快,但由于晶區的限制作用還不足以使晶區的含量發生變化。
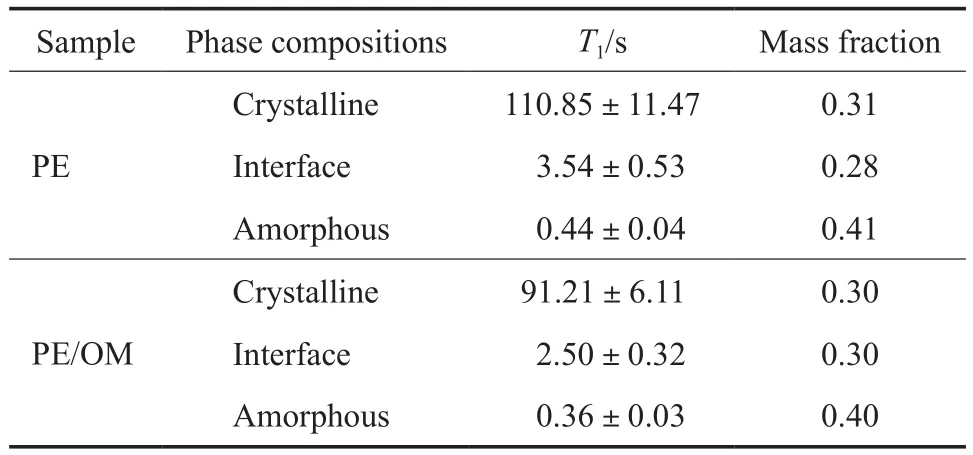
表2 PE和PE/OM的13C T1值Table 2 13C T1 values of PE and PE/OM
3 結論
1)PE的相組成包含晶區、界面區和無定形區,吸附C10H18O至飽和后的PE/OM的晶區含量基本不變,界面區中有很小一部分轉變為無定形區。
2)PE吸附C10H18O至飽和平衡后,晶區、界面區和無定形區的分子鏈運動均變快。
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學生數理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現代企業(2015年9期)2015-02-28 18:56:50
