微反應器內鄰氨基苯甲酸甲酯的連續重氮化工藝
2021-11-03 01:11:04王犇王超尹進華
化工進展 2021年10期
關鍵詞:工藝
王犇,王超,尹進華
(1 青島科技大學環境與安全工程學院,山東 青島 266042;2 青島科技大學化工學院,山東 青島 266042)
現代合成化學在為社會提供有價值產品的同時面臨環境友好性的挑戰,因此,能夠以原子經濟的方式合成復雜結構分子并且具有高化學選擇性的試劑備受關注[1-2]。其中,重氮化合物以其具有多功能性、高效性、清潔性(離去基團為N2)等諸多優點脫穎而出。鄰氨基苯甲酸甲酯(MA)的重氮鹽作為一種典型的重氮化合物被廣泛應用于精細化工、制藥工程等領域,是合成2-(氯磺酰基)苯甲酸甲酯[3]、糖精[4]、法尼基轉移酶抑制劑[5]等多種化合物的重要中間體。
盡管重氮化合物在減少合成步驟和產生廢物方面具有其優越性質,但不能認為它們是安全的或易于處理的。重氮化合物作為高能化合物以其熱不穩定性和爆炸性而聞名,大部分在常溫環境即可自發性分解放熱放氣[6]。同時,其合成過程放熱量大且集中(ΔH介于-150~-65kJ/mol)[7],這對于大型反應設備無疑是危險的。這些反應特性使得工業規模的重氮化生產不得不選用半間歇的加料方式和大功率的低溫冷卻設備來控制產熱速率和移熱速率處于平衡狀態。間歇反應釜傳質傳熱效率低,單位體積換熱面積小,致使較大規模生產仍需要較長反應時間,重氮組分的長時間停留導致平行副反應發生,同時也增加了潛在熱失控風險。
現階段工業生產中MA重氮鹽的合成多選用鹽酸體系,合成路線如圖1 所示。MA 具有胺和酯的雙重性質,難溶于酸且長時間在高濃度酸性體系中會發生一定程度的水解。因此需選用逆法重氮化的合成方式,將MA與亞硝酸鈉溶液混合打漿后滴加到酸中,物料相態的非均一性使得加料過程中物料配比的精準性難以控制。這是該半間歇合成工藝中的嚴重缺陷,影響反應收率的同時增加了反應失控的風險,多起因重氮化合物導致的事故被相繼報道[9-11]。在這種形勢下,尋找一種更穩定、高效、安全的方案,以充分發揮MA重氮鹽和類似重氮化合物在工業有機合成中的巨大潛力,是十分必要和迫切的。
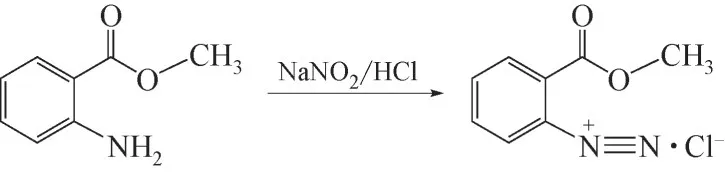
圖1 MA重氮鹽合成方程式
近年來,連續流化學技術發展迅速,微通道反應器作為一種新型反應器,在降低能源消耗[12]、提高傳質和傳熱[13-14]、抑制平行副反應和提高反應體系安全性[15-16]等方面具有獨特優勢。這些特性使得微通道連續流技術廣泛應用于藥物合成、精細化工、生物化學等領域。微通道連續流技術能夠處理多種危險工藝[17-20],重氮化合物的連續合成是現代合成化學領域的熱點問題,最早的芳基重氮鹽組分連續合成方案是由Wootton 等[21]提出,該方案成功地在玻璃集成芯片微通道反應器內實現了苯胺重氮鹽的連續合成,生成的重氮鹽組分進一步與β-萘酚進行連續偶合反應。雖然最終反應收率僅有52%,但為連續重氮化提供了引領性的思路。Pinho 等[22]在微型套管式反應器AF-2400 中安全高效地合成了重氮甲烷,并與甲基化反應、環丙烷反應相結合后成功應用在抗逆轉錄病毒藥物合成中。Yu 等[23]提出了在由重氮化和氟化反應器并聯組成的微通道系統內通過Balz-Schiemann 反應制備芳族氟化物的方法,在重氮反應器內進行了苯胺和2-乙基苯胺的連續重氮化反應,在10~20s 內可獲得較高的產品收率。這表明微通道連續流技術在重氮化合物的合成過程中有著巨大的應用潛力,但芳基重氮鹽組分連續化合成的實際應用還較少。MA重氮鹽合成過程的反應特征與微通道反應器的特征高度吻合,連續流合成技術可能為解決現階段半間歇合成中的問題提供新的視野。
目前報道的微反應器內芳基重氮鹽組分的合成多以可溶性芳胺為主,胺酯雙重性質的難溶性芳胺的連續流合成少有研究,基于上述考慮,本文創新性地提出了MA 重氮鹽連續流合成工藝方案,搭建了相應的微通道反應器裝置。鑒于MA重氮鹽的合成為多變量復雜過程,采用響應面分析法(RSM)[24-25],在單因素實驗的基礎上確定合適的試驗設計因素和水平,通過BBD 中心組合原理構建模型,分析了物料配比、反應溫度、停留時間、流速及各因素交互作用對連續重氮化收率的影響,確定了微通道反應器內合成MA 重氮鹽的最優工藝條件。為比較該合成工藝與傳統半間歇工藝在改善工藝條件、提高反應安全性和降低能耗等方面的效果,在10L 反應釜中建立了特定的半間歇合成實驗及響應面分析。該研究為微反應器內重氮化合成技術的工業化提供了數據基礎和技術支持。
1 實驗
1.1 試劑和儀器
鄰氨基苯甲酸甲酯(99%),阿拉丁試劑(上海)有限公司;鹽酸(37%),煙臺遠東精細化工有限公司;亞硝酸鈉、1-萘酚、氫氧化鈉、氨基磺酸,分析純,國藥集團化學試劑有限公司;硼砂,分析純,天津致遠化學試劑有限公司。
紫外-可見分光光度計,UV-2600 型,島津實驗器材有限公司;三重四極桿氣質聯用儀,7890B/7000C 型,安捷倫科技有限公司;10L 雙層夾套玻璃反應釜,上海科興儀器有限公司;FP40-MA 加熱制冷循環浴槽,Julabo 技術有限公司;PL602E型分析天平,梅特勒-托利多國際貿易有限公司;常規玻璃儀器。
Advanced-FlowTM高通量微通道反應器系統(美國Corning公司)由特種玻璃模塊區、物料輸送區、熱量交換區、安全控制區和相關連接部件組合而成。AFRTM-G1 反應模塊作為反應載體,其結構如圖2 所示。反應器由玻璃模塊和連接模塊組成,玻璃模塊被四層玻璃分隔成三個中空空間。兩端為兩個熱交換層,持液量14mL;中間為反應層,持液量為8.2mL。反應通道由一系列內徑為1mm的微型“心形結構”組成,具有完全混合流動特性,這種結構特征使其單位換熱面積可達2500m2/m3。
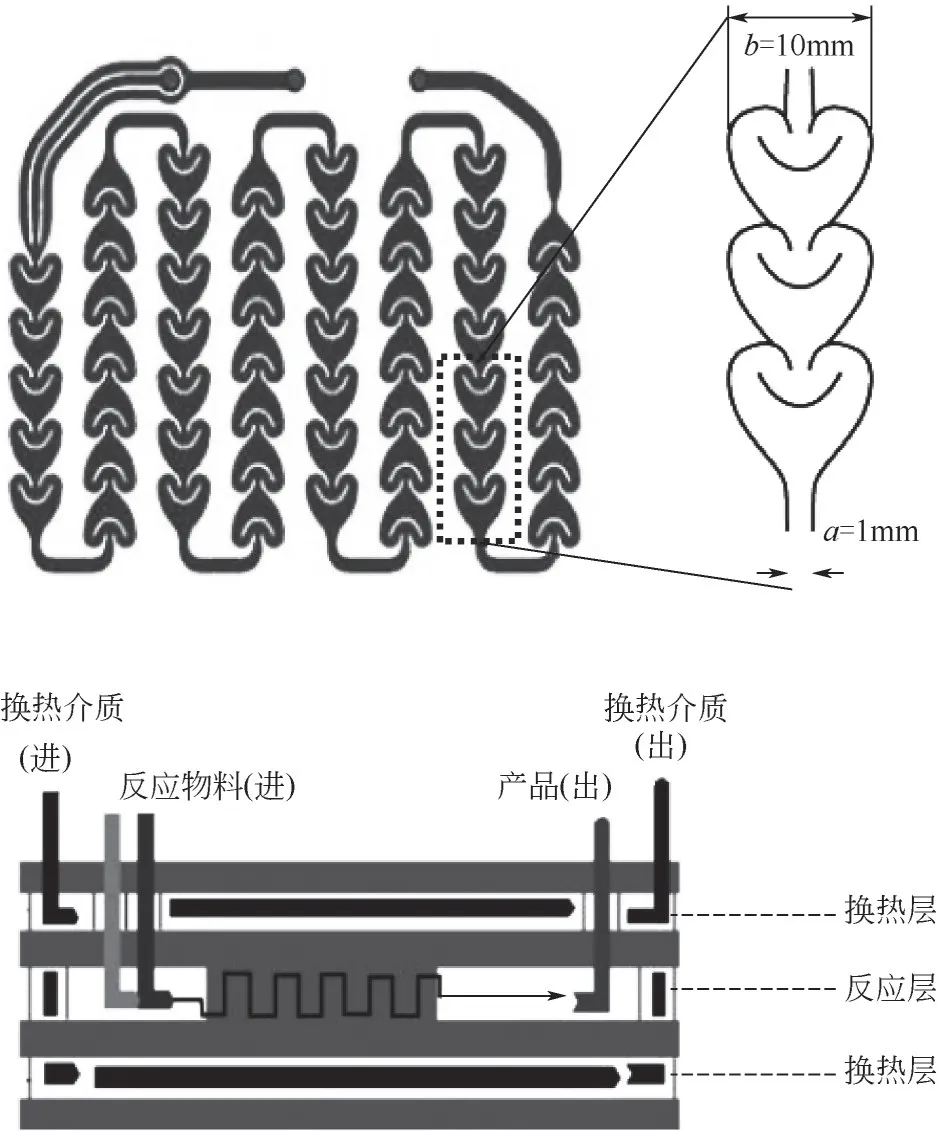
圖2 微通道混合脈沖結構
1.2 實驗流程
1.2.1 半間歇合成工藝流程
將MA和25%的亞硝酸鈉溶液按所需摩爾比充分混合,攪拌打漿至呈乳化液狀態。取所需用量的25%鹽酸溶液(預冷)置于玻璃反應釜中,滴加MA和亞硝酸鈉溶液的混合乳化液,控制反應溫度和加料速度(加料過程控制在15min),滴加完畢后繼續反應15min,向體系中加入少量氨基磺酸去除過量的亞硝酸,抽濾除去反應體系中的焦油狀雜質,所得棕黃色澄清溶液即為MA重氮鹽溶液。
1.2.2 微通道連續流合成工藝流程
微通道連續流重氮化反應裝置如圖3所示,利用恒溫循環設備控制系統溫度,待溫度達到設定值并穩定,根據實驗需求設定流速,物料泵同時向微通道反應器輸送物料。物料1(25%的鹽酸溶液)經泵5泵入預處理區進行預冷或預熱,物料2(MA)和物料3(25%的亞硝酸鈉溶液)進入預處理區進行預混合。兩股物料預處理后進入反應區進行反應,在反應淬滅區內由物料4(氨基磺酸溶液)去除過量的亞硝酸,棄去前50mL 的反應混合物。通過調節計量泵的流量控制反應物料的摩爾比,通過調節反應區的模塊數量來控制反應液的停留時間。
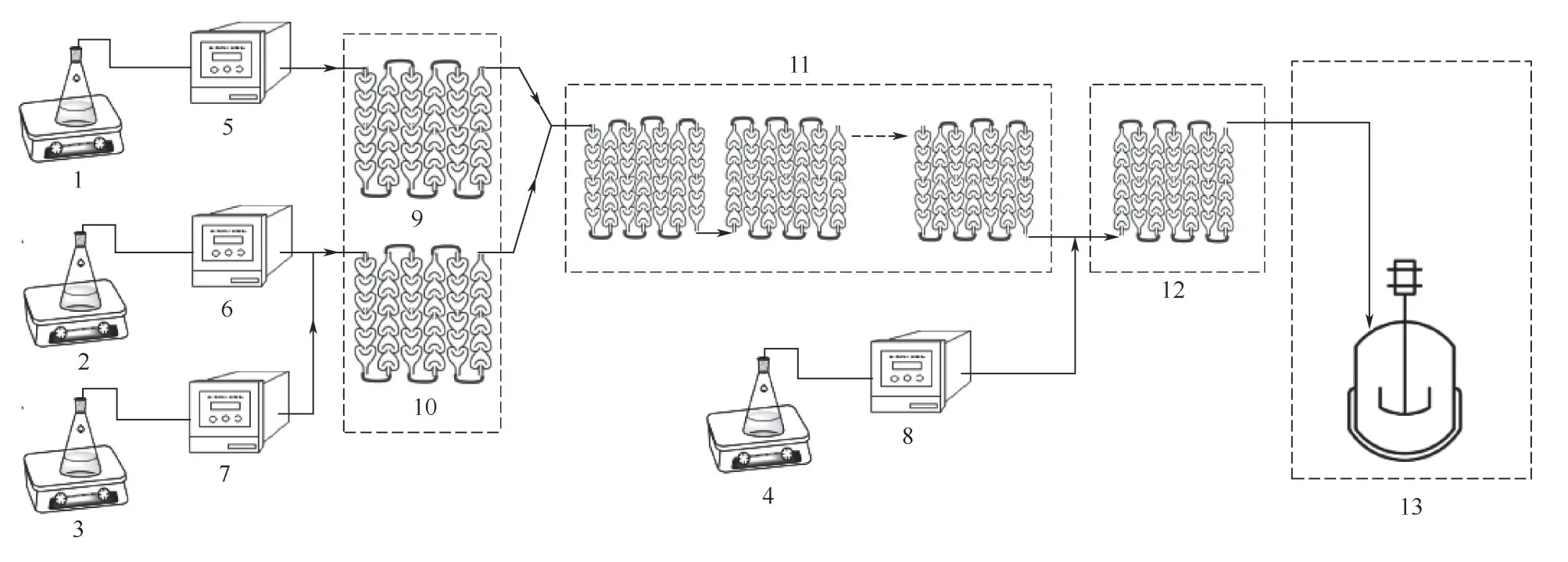
圖3 微通道連續流重氮化反應的實驗裝置
1.3 分析方法
參照文獻[26]報道的方法,根據重氮化反應在低濃度的強酸溶液中反應比較完全且穩定,重氮鹽在堿性條件下與1-萘酚迅速偶合顯色的原理,采用分光光度法定量分析MA重氮鹽的濃度,計算重氮化收率。具體操作步驟如下:取MA 標準品0.500g,加入0.1mol/L的鹽酸50mL溶解,以水為溶劑稀釋至50mg/L 記作標準溶液;從標準溶液中分別移取0.5mL、1.5mL、3mL、5mL、7.5mL于25mL容量瓶,依次加入0.1mol/L 的鹽酸6mL、50g/L 亞硝酸鈉0.5mL、pH=12.5的硼砂-氫氧化鈉緩沖溶液10mL、1-萘酚乙醇水溶液1mL,加水定容(加入鹽酸、亞硝酸鈉溶液、緩沖劑、顯色劑的過程都記作稀釋),放置15min 后于490nm 處測定吸光度,取3 次測量值,以濃度和對應吸光度繪制標準曲線。擬合所得標準曲線為A=0.15032C+0.06586,R2=0.9921,表明在1~15mg/L 的范圍內濃度與吸光度呈較好的線性關系。
取1mL 待測樣品急冷后定容至1000mL 記作溶液A,取1mL溶液A置于25mL容量瓶,加入6.5mL水、pH=12.5 的硼砂-氫氧化鈉緩沖溶液10mL、1-萘酚乙醇水溶液1mL,加水定容。取3次測量所得平均吸光度,與標準曲線對比計算已轉化為MA重氮鹽的MA的量。通過式(1)計算重氮化收率(Y)。
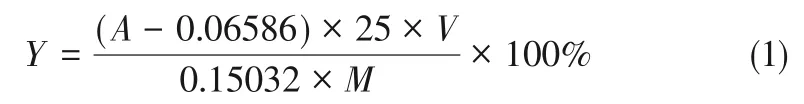
式中,A為3 次測量所得平均吸光度;V為重氮液體積,mL;M為生成VmL 重氮溶液所消耗的MA的質量,mg。
2 單因素實驗結果與討論
為確定合適的響應面試驗設計因素和水平,以MA重氮鹽的收率為考察指標,對反應物摩爾比、反應溫度、反應時間、流速進行單因素實驗考察。標準實驗條件為半間歇合成中物料摩爾比n(MA)∶n(亞硝酸鈉)∶n(鹽酸)=1∶1.3∶3、反應溫度15℃、反應時間15min,微通道連續流合成中物料摩爾比為n(MA)∶n(亞硝酸鈉)∶n(鹽酸)=1∶1.1∶2.6、計量泵1 流速為23.2g/min、計量泵2 流速為9.2g/min、計量泵3流速為18.6g/min、反應溫度為35℃、停留時間為40s。當研究不同的反應條件時,所研究的條件被改變,其他條件保持不變。
2.1 亞硝酸鈉用量對反應的影響
重氮化反應中的有效重氮化劑是由亞硝酸鈉和鹽酸反應生成的亞硝酸,理論MA與亞硝酸鈉的摩爾比應為1∶1,但亞硝酸的不穩定性使得在實際合成過程中亞硝酸鈉用量高于理論值,需根據反應體系的特性來選擇適宜的摩爾比。如圖4所示,在半間歇合成過程中,隨著MA與亞硝酸鈉摩爾比的增加,MA重氮鹽的收率呈持續上升趨勢,直至比理論值高出30%后呈平穩趨勢。該趨勢并不只是亞硝酸不穩定的單一因素所導致,物料相態的非均一性使得加料過程中物料配比的精準性難以控制,階段性的亞硝酸鈉過高或過低均會對反應造成負面影響。在微通道連續流工藝中,MA和亞硝酸鈉溶液分別由計量泵泵入預混模塊,在毫米級混合結構下充分混合后進入反應區,對物料初始混合的強化和混合與反應過程連續進行,保證了物料的精準配比。因此,微通道工藝所需的亞硝酸鈉用量較理論值高出10%時,重氮化收率就達到了90.3%且遠高于半間歇工藝。
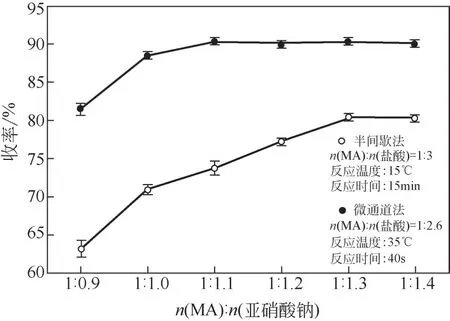
圖4 MA與亞硝酸鈉摩爾比對重氮化收率的影響
2.2 鹽酸用量對反應的影響
理論上1mol MA 完全重氮化需要2mol 的酸,在實際合成過程中,重氮反應體系需保持一定的酸度以抑制副反應和維持重氮鹽組分的穩定性。如圖5 所示,半間歇工藝中,隨著MA 與鹽酸摩爾比的增加,重氮化收率上升趨勢明顯。當MA與鹽酸的摩爾比為1∶3 時,重氮化收率達到峰值。在半間歇合成過程中,反應液中有明顯焦油狀物質生成,且鹽酸用量越低該物質的含量越多。微通道連續流工藝中MA與鹽酸的摩爾比為1∶2.6時,反應收率到達峰值趨于平穩,而且反應液顏色正常,未觀察到焦油狀物質生成的現象。這一趨勢得益于微通道反應器無反混、持液量小的特性。生成的重氮組分能被及時地移出進入后續反應,不需要大量的酸來抑制平行副反應。
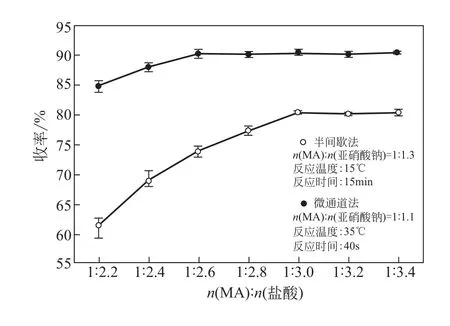
圖5 MA與鹽酸摩爾比對重氮化收率的影響
2.3 反應溫度對反應的影響
重氮化合成過程因其強放熱性和體系多組分熱不穩定性使得反應溫度成為最值得關注的工藝參數。如圖6所示,隨著溫度的升高,兩種合成方式的收率都呈先上升后下降的趨勢,低溫時反應未被充分引發,表現出低活性,高溫下反應快速進行的同時平行副反應和亞硝酸分解速率也被加劇。在半間歇工藝中,15℃時重氮化收率達到峰值80.4%。值得注意的是,在大于25℃的半間歇實驗中均出現了階段性大幅超溫現象,導致重氮化收率大幅下降并伴隨大量焦油狀物質生成。間歇反應釜的固有特性和加料方式決定了反應體系局部溫度過高產生“熱點”現象,反應釜內熱量累積易引發重氮組分的二次分解放熱導致反應失控,這在傳統的半間歇生產中是無法避免的。在微通道合成過程中,重氮化反應被允許以高的反應溫度、短的停留時間以獲取更高的重氮化收率。當反應溫度為35℃,MA重氮鹽的收率可達90.3%。同時,微通道反應器具有極高的熱慣性因子,即使反應過程發生偏差,體系的溫升也只是極小值,不會引發反應的熱爆炸效應[27],為高溫條件下進行重氮化反應提供了可行性和安全基礎。
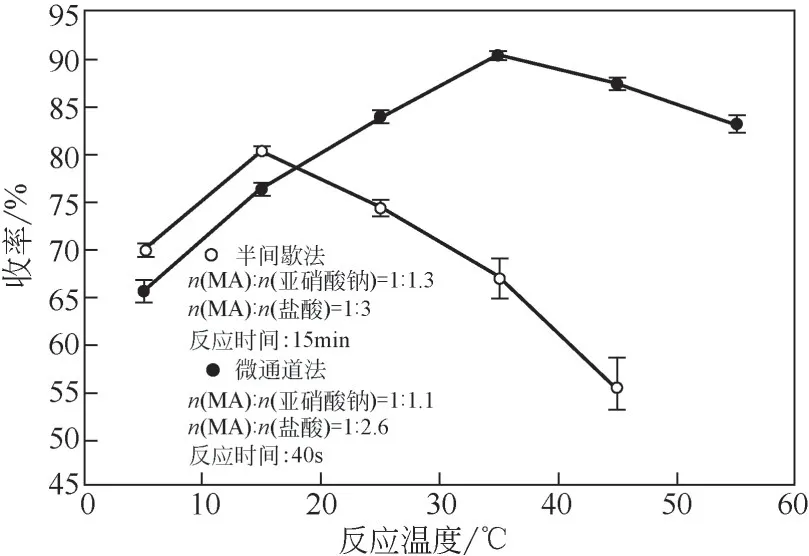
圖6 反應溫度對重氮化收率的影響
2.4 停留時間對反應的影響
適當的反應時間不僅可以保障產品的高質量,而且可以提高生產效率。如圖7所示,半間歇釜內物料傳質效果差,反應完全需要15min。微通道模塊中反應液在“心形結構”擾動下,強化了混合,分子動能可支持分子間的快速碰撞,停留時間為40s 時重氮化收率趨于平穩,相比半間歇工藝極大縮短了反應時間。但停留時間過短,反應不能充分進行,物料進入反應淬滅區后,部分未參與反應的亞硝酸在氨基磺酸的作用下被去除,加劇MA重氮鹽和剩余芳胺組分的自偶合現象,導致重氮化收率的降低。
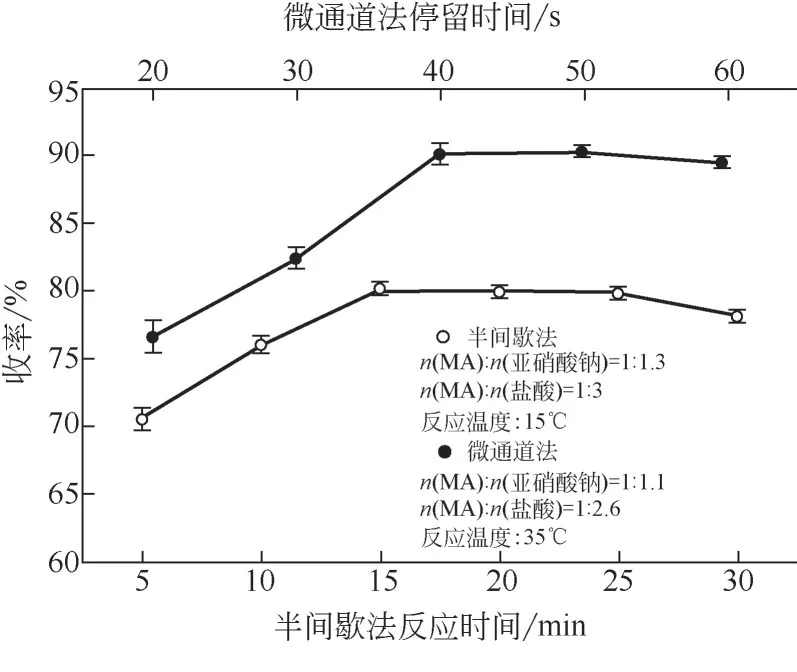
圖7 反應時間對重氮化收率的影響
2.5 流速對反應的影響
在微通道反應器中,流速是影響混合和傳質效果的決定性因素。在標準實驗條件下,保持反應停留時間不變,通過改變反應區模塊串聯數量調節反應體系流量考察了流速對重氮化收率的影響,結果如圖8 所示。G1 模塊中“心形結構”單元內部障礙和沿流動路徑不斷變化的橫截面造成了反應通道內的壓力變化,可擠壓液體破裂成小尺寸液滴以增強兩相混合效果和提供高效的傳質界面面積。但反應體系流速較低時,這種效果將被極大削弱,導致在25.5~51g/min的流速區間時,反應體系未能達到最佳的液滴尺寸分布和傳質效果,重氮化收率隨流量的增加呈現上升趨勢。當反應體系流量大于51g/min 時,重氮化收率趨于穩定,表明此時反應體系已經達到最佳混合狀態,基本消除了傳質過程對反應的影響。
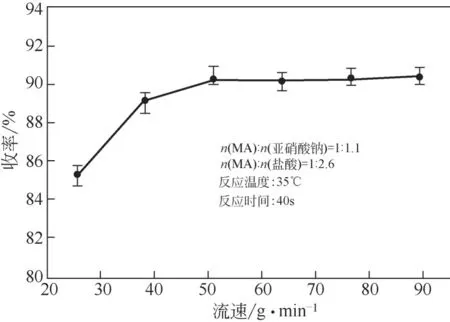
圖8 流速對重氮化收率的影響
3 響應面優化實驗設計與討論
3.1 中心組合實驗設計及結果
由單因素實驗結果可以看出MA的重氮化反應為多變量復雜過程,為分析兩種合成工藝中各因素交互作用對反應收率的影響,進而確定最佳工藝參數。根據BBD 中心組合原理,以MA 重氮鹽收率(Y)為響應值,以MA 與亞硝酸鈉摩爾比(A)、MA與鹽酸摩爾比(B)、反應溫度(C)、反應時間(D)為響應因素,在單因素實驗得出的中心實驗點的基礎上,對連續流和半間歇合成工藝均采用Design Expert 8.0.6軟件進行四因素三水平的響應面實驗設計,所選擇的因素及水平取值見表1和表2,所得29組實驗點(24組析因點、5組中心點)及結果見表3。
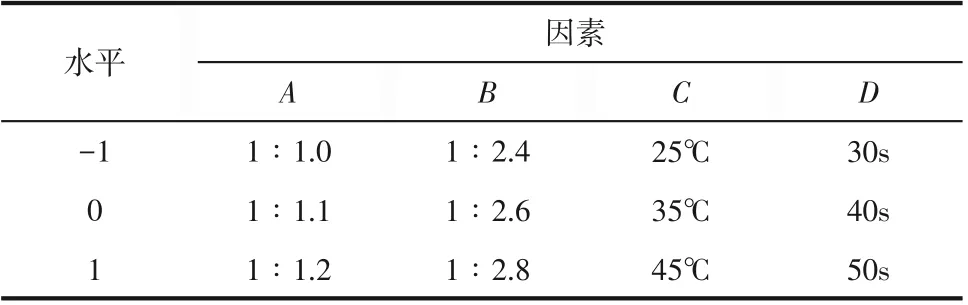
表1 微通道連續流實驗因素及水平
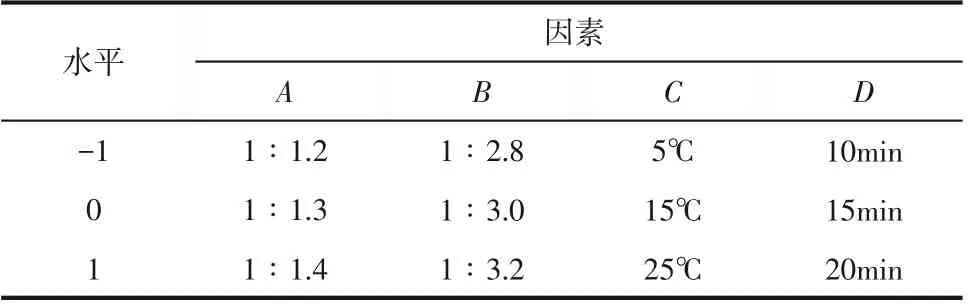
表2 半間歇實驗因素及水平
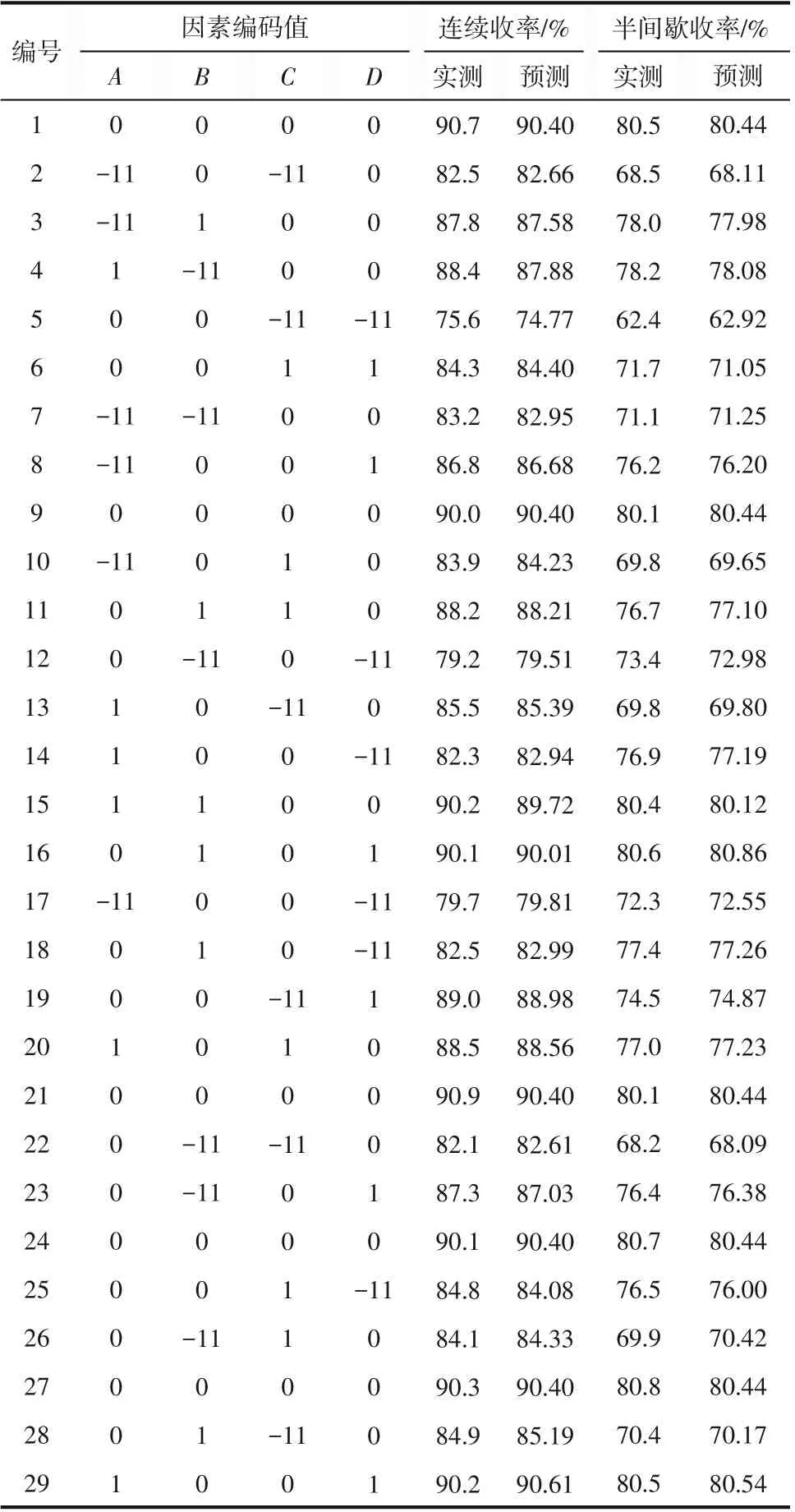
表3 實驗設計矩陣及響應值
3.2 響應面實驗模型構建及顯著性檢驗
采用Design Expert 8.0.6 軟件對擬合模型(線性函數,2FI 模型、二階模型、三階模型)的顯著性、失擬項、相關性數據進行對比,二階模型獲得了最低的標準偏差以及最高的確定系數(R2)、調整系數(RAdj2)和預測系數(RPred2)。因此,選取二階模型對各因素及其響應值進行回歸擬合,其數學模型如式(2)所示。
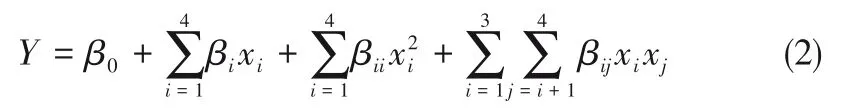
式中,Y為預測響應值;β0為常量系數;βi為線性效應系數;βii為二次效應系數;βij為交互效應系數。
根據響應變量的編碼值獲得的MA重氮鹽收率回歸方程如式(3)、式(4)所示。
連續流工藝MA重氮鹽收率回歸方程
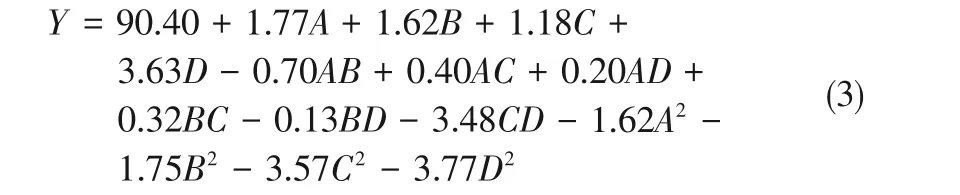
半間歇工藝MA重氮鹽收率回歸方程
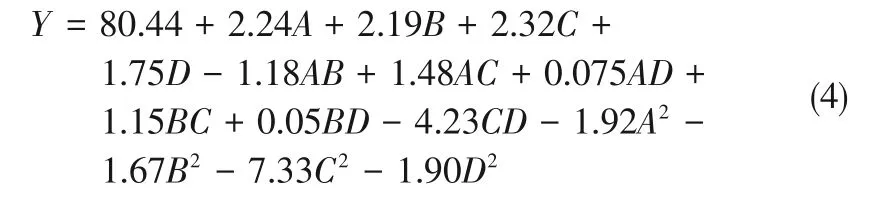
進一步對所得的二次多項回歸模型進行方差分析(ANOVA)。F值及其對應的P值被視為檢驗相關系數顯著性的標準[28],在該分析中,考慮了5%的顯著性水平,意味著如果P值低于5%,則模型是顯著的(P值低于1%視為極顯著)。由表4、表5 可知,兩種合成工藝的響應面整體模型P值均小于0.0001,意味著模型是極顯著的,失擬誤差的P值分別為0.2240 和0.2385,均不具有顯著性,表明回歸方程與實測值有極高的擬合精度。
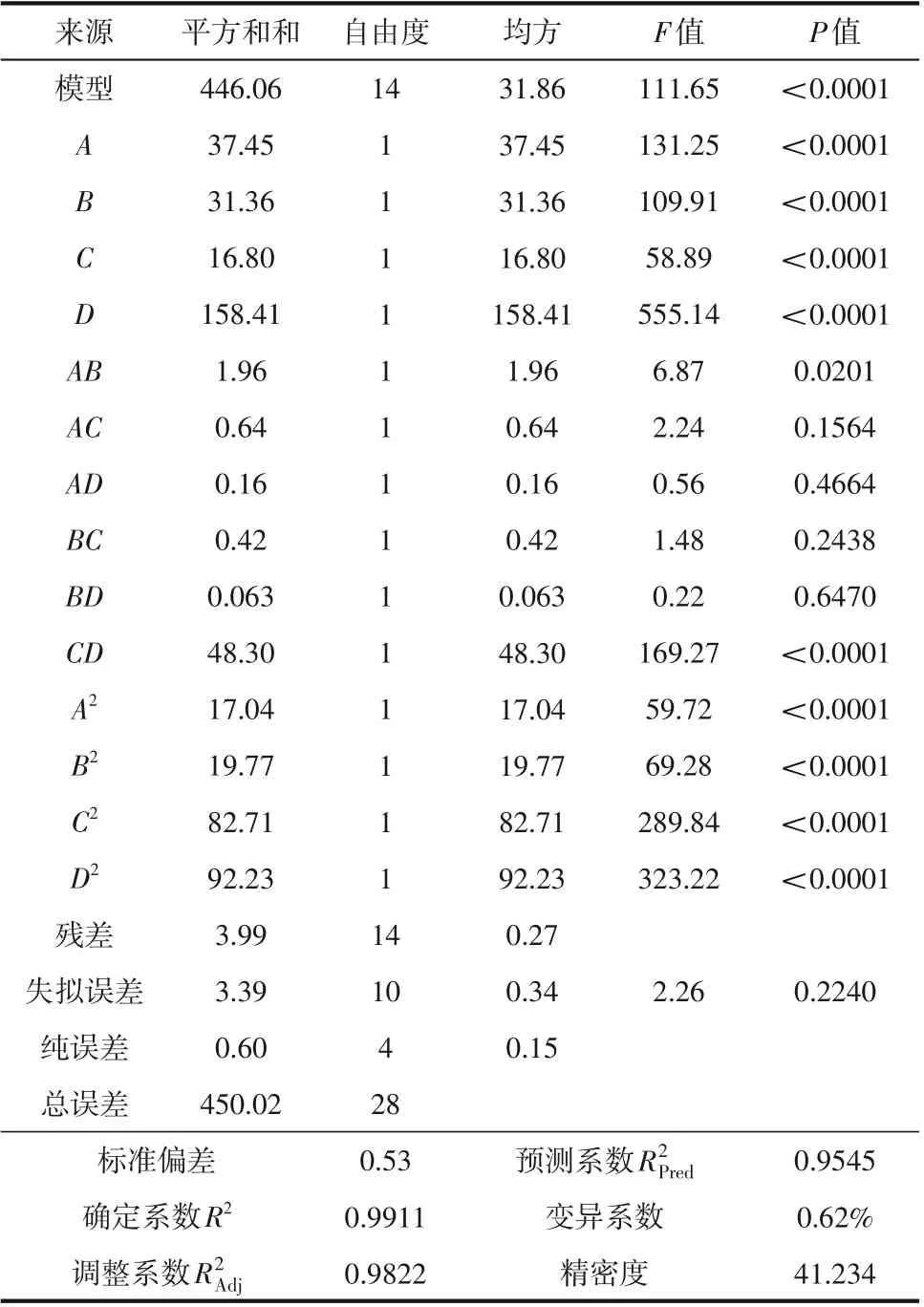
表4 連續流重氮化回歸方程方差分析和顯著性檢驗結果
各回歸系數的顯著性檢驗表明,兩模型中的一次項(A、B、C、D)的線性效應和二次項(A2、B2、C2、D2)的交互效應均表現為極顯著。連續流回歸模型中AB項交互效應顯著,CD項交互效應極顯著;半間歇回歸模型AB、AC、BC、CD項的交互效應極顯著,表明各因素對響應值的影響并非單一的線性關系。根據模型F值可知,連續流合成工藝各因素對重氮化收率的影響強弱次序為反應時間>亞硝酸鈉用量>鹽酸用量>反應溫度,半間歇合成工藝各因素對重氮化收率的影響次序為反應溫度>亞硝酸鈉用量>鹽酸用量>反應時間。不同因素及其交互作用對響應值的影響將在3.4 節中詳細介紹。
3.3 二次多項回歸模型驗證
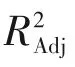
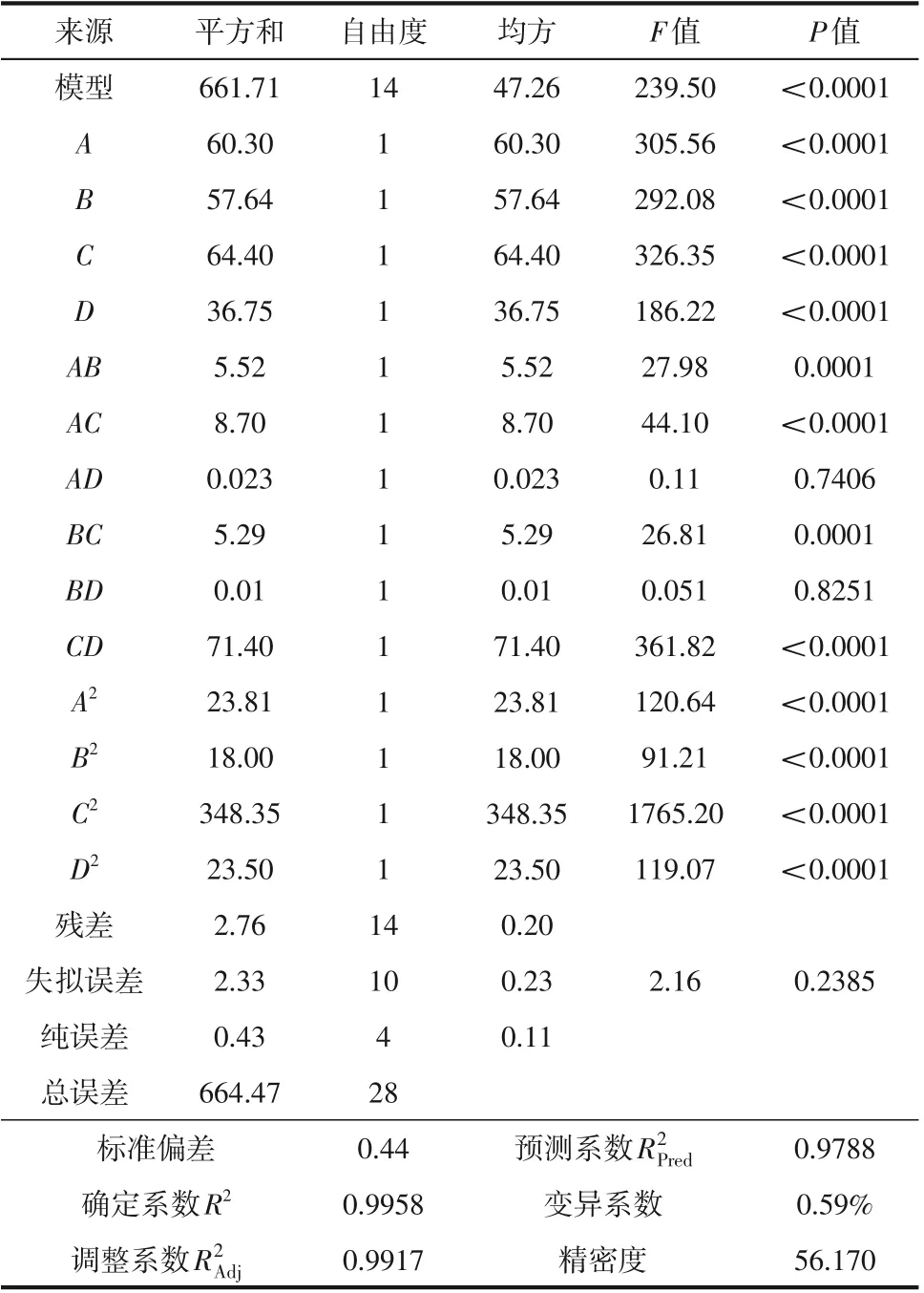
表5 半間歇重氮化回歸方程方差分析和顯著性檢驗結果

圖9 殘差正態概率圖
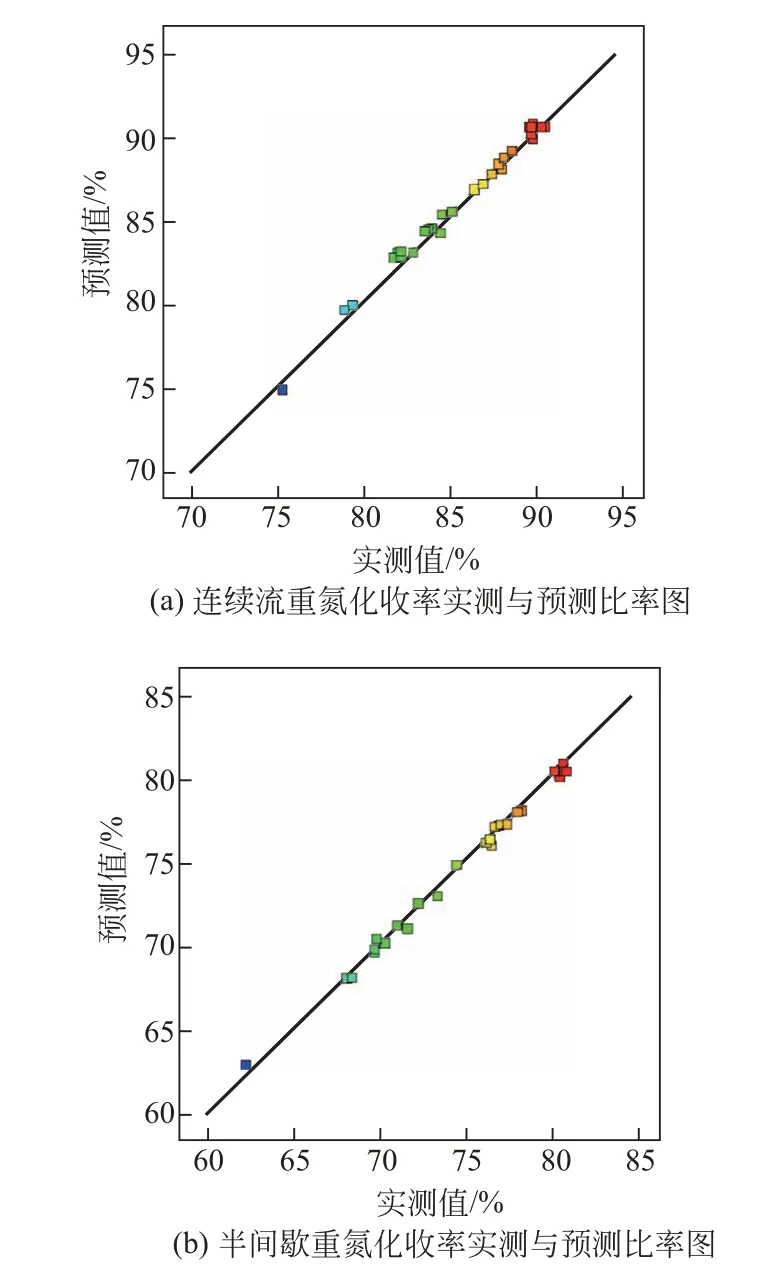
圖10 實測與預測比率圖
3.4 不同因素及其交互作用對反應的影響
保持兩個因素固定在中心值不變,考察其他兩個因素對響應值的影響,構建了兩種合成方法二次回歸模型中AB、AC、BC、CD項交互效應對重氮化收率影響的3D響應面曲圖及2D等高線圖,結果如圖11~圖14所示。3D曲面的傾斜程度和等高線的形狀可以直觀反映出各因素交互效應對響應值的影響,傾斜度越大,該因素對響應值的影響越顯著;等高線形狀越趨近于圓形,兩因素交互效應越弱[30]。
如圖11 所示,等高線圖左下部分密集且對應區域的3D 曲面傾斜程度大,表明亞硝酸鈉和鹽酸用量均不足時對重氮化收率影響顯著,該條件下兩因素存在交互效應。但值得注意的是,圖11(a)所示的等高線圖相較于圖11(b)更趨近于圓形,雖然半間歇工藝使用了高劑量的亞硝酸鈉和鹽酸,但兩因素的交互效應對重氮化收率的消極影響仍強于連續流工藝。
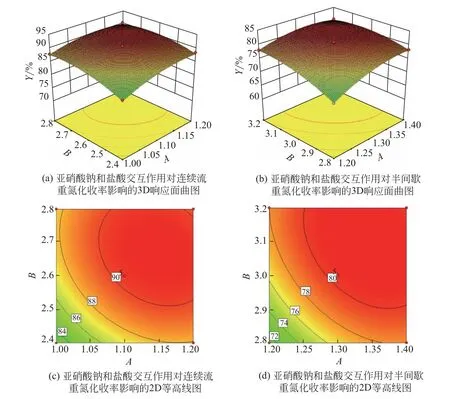
圖11 亞硝酸鈉和鹽酸交互效應對重氮化收率影響
圖12 顯示了反應溫度與亞硝酸鈉用量的交互效應,兩種合成工藝的響應面圖表現出明顯差異。半間歇工藝構建的3D 曲面在整個反應溫度范圍內呈現拋物線形傾斜結構。反應溫度與亞硝酸鈉用量之間存在著極顯著的交互效應,這歸因于亞硝酸的熱不穩定性,未及時參與反應的亞硝酸在高溫下分解。連續流工藝構建的3D 曲面平滑且等高線圖趨近于圓形,表明反應溫度與亞硝酸鈉用量在所選參數區域內無顯著的交互效應。該趨勢得益于高溫下反應物具有高反應活性,在短時間內即可完成向產物的轉換,大幅降低了亞硝酸在高溫下長時間停留導致的損耗。
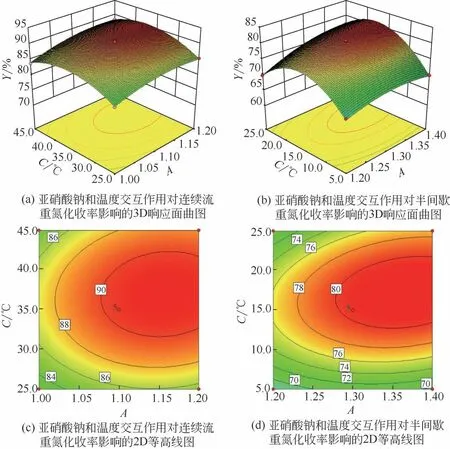
圖12 亞硝酸鈉和反應溫度交互效應對重氮化收率影響
圖13 與圖12 表現出相同的變化趨勢,可作類似分析。MA重氮鹽的穩定性同時受到反應溫度和體系酸度的影響,這決定了反應溫度與鹽酸用量之間存在著交互效應,由圖13(d)可知,等高線沿C軸方向變化密集,明顯高于B軸方向,這意味著半間歇合成中反應溫度與鹽酸用量交互效應顯著且反應溫度對MA 重氮鹽收率的影響起到了主導作用。相比半間歇工藝,連續流工藝能及時移除重氮組分的特性將,高溫對反應帶來的負面影響降到了較低的水平,即使低劑量的鹽酸也足以保持重氮組分在反應時間內相對穩定。
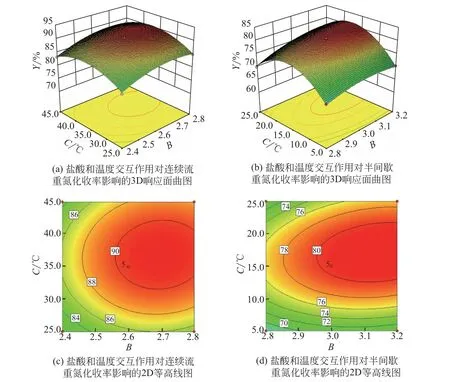
圖13 鹽酸和反應溫度交互效應對重氮化收率的影響
相比于連續流工藝中其他因素的交互作用,反應溫度和停留時間的交互效應對重氮化收率的影響表現出復雜的變化趨勢。如圖14(a)所示,3D 曲面呈現極大的傾斜程度,25℃時反應物具有的反應活性并不能保證反應在20s內充分完成,未反應完的原料在淬滅區內發生大量副反應,MA重氮鹽的收率僅為74.77%。升高溫度和延長停留時間均使得反應趨于完全,但這種趨勢在過高的溫度和過長停留時間的條件下將被逆轉。半間歇工藝構建的3D曲面在C軸兩端對應的整個時間范圍內呈現明顯的區域性凹陷,而連續流工藝構建的3D 曲面中區域性凹陷出現在低停留時間對應的整個溫度范圍。這些結果表明,連續流合成工藝有效解決了傳統半間歇合成過程中重氮體系對溫度的高敏感性和低溫的依懶性,在一定程度上實現了MA重氮鹽收率由反應溫度主導向反應時間主導的轉變。因此,對于連續化合成工藝來說,合適的停留時間是關鍵,這也與表4中方差分析的結果一致。
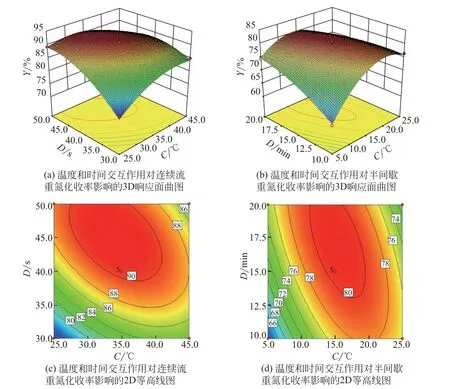
圖14 反應溫度和時間交互效應對重氮化收率影響
3.5 最佳合成參數及驗證
根據回歸模型求解得到兩種工藝最佳合成條件。微通道連續流工藝最佳合成條件為n(MA)∶n(亞硝酸鈉)∶n(鹽酸)=1∶1.15∶2.67、反應溫度34.62℃、停留時間45.07s,預測此條件下MA重氮鹽收率為92%。半間歇工藝最佳合成條件為n(MA)∶n(亞硝酸鈉)∶n(鹽酸)=1∶1.35∶3.11、反應溫度16.73℃、停留時間16.34min,預測此條件下MA重氮鹽收率為82%。
在模型預測的最佳條件下進行3組平行驗證實驗,微通道連續流工藝重氮化收率平均值為92.14%,半間歇工藝重氮化收率為81.35%,與預測值相比相對誤差僅為0.14%和0.65%,表明預測模型擬合性能高,在最佳工藝條件下反應穩定,有較好的重現性。
3.6 微通道連續流工藝長期運行及工業放大可行性
在響應面優化所得的最佳工藝條件下,對MA連續重氮化工藝長期運行的可行性進行初步驗證。在連續20h的運行過程中,反應體系保持穩定,未出現局部沉淀、通道堵塞等異常現象。以2h 為周期進行取樣分析,MA 重氮鹽收率均穩定在92%±0.3%,表明該工藝可長期平穩運行,適用于工業化生產。
與工業規模的釜式反應器相比,單組AFRTMG1 微通道反應器的產能僅可滿足小規模合成需求(表6)。但可模塊式規模化是微連續流技術的一個重要優勢,通過并行增加微反應器的數量以擴大產量規模和提高使用靈活性是可行的有效方式。微反應器的關鍵優勢是源于其固有的小尺寸特性,“數增放大”的方式避免了傳統半間歇工藝需要通過“小試-中試-生產”逐級尺寸放大而導致傳質傳熱等方面的放大效應,使得在大規模工業化生產中也可發揮其全部潛力。值得關注的是,多通道間流量分配的均勻性是“數增放大”所面臨的關鍵問題。近年來多種流體進口分配技術的提出為解決該問題提供了有效方案[31-33],極大地促進了微化學技術從基礎研發到工業化應用的轉變。在“數增放大”方式和流量分配技術的支持下,目前已實現由AFRTM-G1 研發的小規模合成方案無縫放大到AFRTM-G4 直接用于工業化生產[34],并且擁有2000 噸的年產量可滿足工業規模的需求。

表6 Corning AFRTM微通道反應器與半間歇反應釜放大數據對比
4 重氮化反應副產物初步討論與表征
在以上多組實驗中均觀察到反應體系內出現了明顯的焦油狀物質,嚴重影響產品質量,而且從重氮體系中去除這些副產物的過程也存在危險。基于該現象,對MA重氮鹽合成過程中的副反應進行了探討。
采用氣相色譜-質譜法(GC-MS)對該物質進行了分析,經過GC 的分離得到4 種組分。EI 質譜譜圖如圖15所示,4種組分的分子離子峰質荷比分別為153、170、138、156。通過解析和NIST 譜庫檢索對質譜圖進行了分析,確定4種組分分別為鄰羥基苯甲酸甲酯(a)、鄰氯苯甲酸甲酯(b)、鄰羥基苯甲酸(c)、鄰氯苯甲酸(d)。結果表明,該物質是由MA 重氮鹽經多步分解產生的混合物。[R—N+≡≡N]帶有一個單位正電,與其相連的碳原子有較低的電子云密度,因此反應體系中的親核試劑[OH-]、[Cl-]在與[R—N+≡≡N]相連的碳原子上易發生親核取代反應,如圖16所示(Nu為親核試劑)。
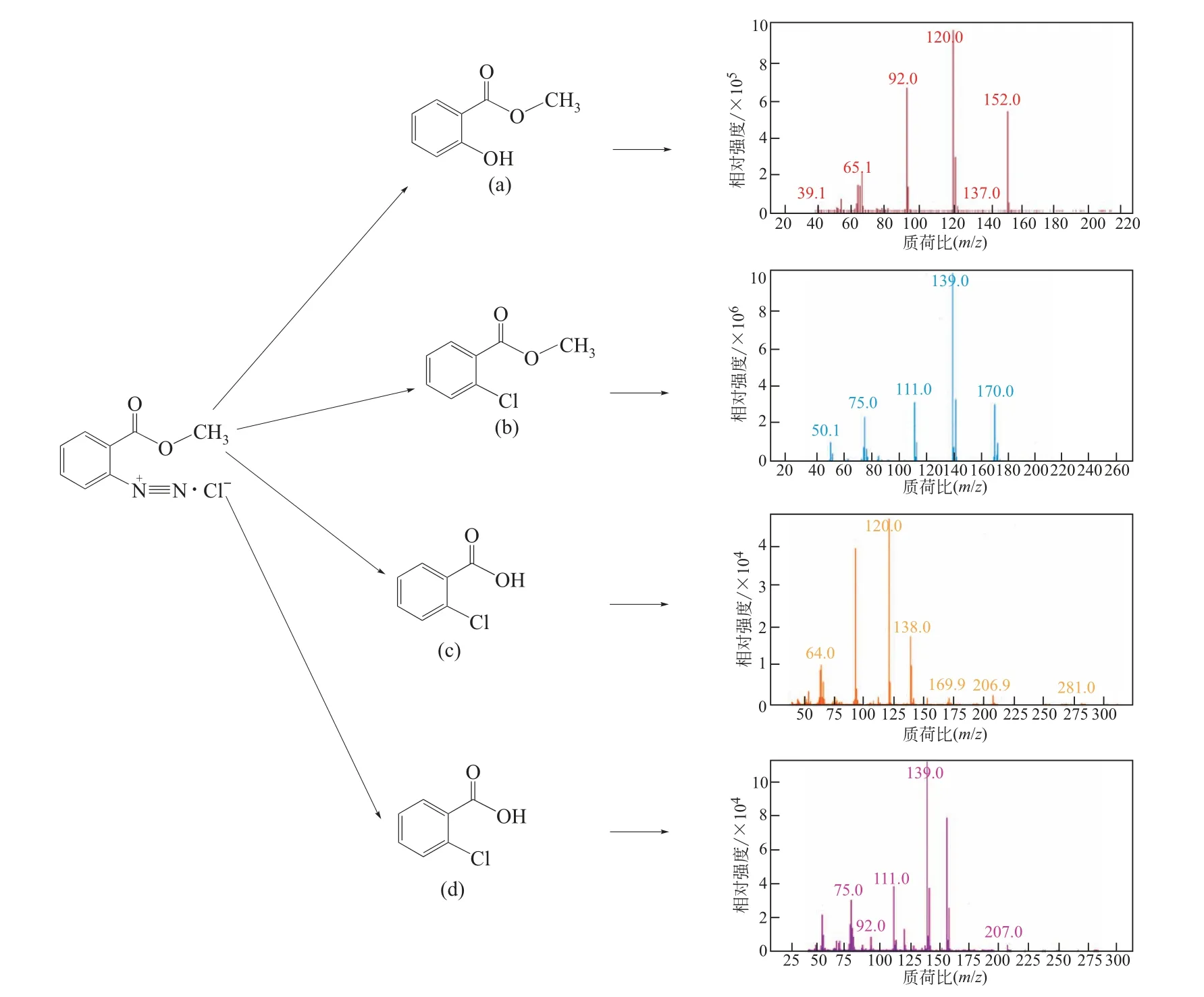
圖15 焦油狀副產物的組成及EI質譜譜圖
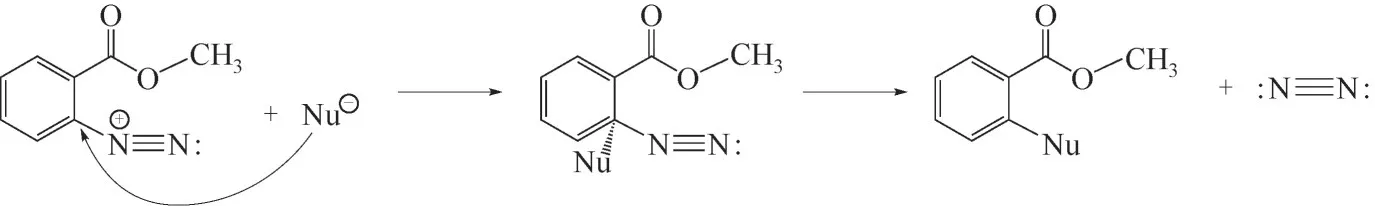
圖16 重氮基親核取代反應
半間歇工藝中因為反應釜的特性,即使在經響應面優化后的工藝條件下,反應體系內仍會有焦油狀物質生成,這些組分需要過濾除去后才能投入后續反應。優化后連續流工藝合成的MA重氮鹽溶液均未觀察到焦油狀物質的生成,該重氮組分可直接用于后續反應,極大地提高了生產效率和避免了從重氮組分中分離雜質所帶來的風險。
5 結論
本文成功開發了微通道反應器內重氮化反應制備MA重氮鹽的穩定連續流工藝。與傳統半間歇合成工藝相比,這種新的合成方法允許在降低工藝危險性的同時以更高的產品收率和生產效率支持反應。響應面分析結果表明,相比于半間歇合成工藝,連續流合成工藝大幅降低了各因素之間的交互效應對反應的負面影響,使反應更加穩定可控且資源被最大化利用。優化后的連續流工藝最佳反應條件為n(MA)∶n(亞硝酸鈉)∶n(鹽酸)=1∶1.15∶2.67、反應溫度34.62℃、停留時間45.07s,此條件下MA重氮鹽收率為92%,產物無雜質可直接用于后續反應。半間歇合成工藝在最佳條件下收率僅有82%且產物中含有因重氮組分分解產生的雜質。
微通道連續流技術有效解決了MA半間歇重氮化工藝中的固有缺陷和重氮化合成體系對溫度的高敏感性以及低溫的依賴性,在一定程度上實現了重氮化收率由反應溫度主導向反應時間主導的轉變。“數增放大”的方式可支持在不喪失微反應器固有小尺寸的特性下實現無縫規模化放大以滿足工業需求。該工藝可作為一種本質安全化的生產方式,具有良好的工業應用前景,有望為類似MA重氮鹽的其他危險物質的合成提供一條可工業化應用的途徑。
猜你喜歡
中國特種設備安全(2022年5期)2022-08-26 09:19:32
礦產綜合利用(2020年1期)2020-07-24 08:50:40
山東冶金(2019年6期)2020-01-06 07:45:54
收藏界(2019年2期)2019-10-12 08:26:06
世界農藥(2019年2期)2019-07-13 05:55:12
世界農藥(2019年2期)2019-07-13 05:55:10
模具制造(2019年3期)2019-06-06 02:11:00
山東工業技術(2016年15期)2016-12-01 05:30:59
銅業工程(2015年4期)2015-12-29 02:48:39
新疆鋼鐵(2015年3期)2015-11-08 01:59:52
