氣相色譜-串聯質譜法測定豆乳中12 種有機磷及氨基甲酸酯類農藥殘留
2021-11-01 01:54:02謝瑞龍梁建英
乳業科學與技術 2021年5期
謝瑞龍,梁建英
(內蒙古伊利實業集團股份有限公司,內蒙古 呼和浩特 010110)
有機磷和氨基甲酸酯類殺蟲劑因具有廣譜、高效、廉價等優點,已被廣泛用于農業生產的蟲害防治過程中。但由于這些農藥的不合理使用,不但對生態環境造成破環,藥物殘留還直接威脅到人類的身體健康[1-2]。 根據作用機制,這2 類農藥會抑制動物或人體內乙酰膽堿酯酶的活性,造成乙酰膽堿不斷累積,影響其正常傳導,導致中毒現象的發生;若在人體內長期蓄積還會致畸、致癌、致突變,甚至死亡[3-5]。
豆乳作為植物乳的一種,因其含有豐富的營養成分,有助于調節內分泌、預防骨質疏松癥、更年期綜合征以及心血管疾病等,所以越來越受到消費者 喜愛[6-7]。目前,豆乳中有機磷及氨基甲酸酯類農藥檢測方法少有文獻研究,而植物源性食品的檢測方法有酶抑制法[8]、酶聯免疫法[9]、氣相色譜法[10-11]、液相色 譜法[12-13]、氣相色譜-質譜法[14-15]、氣相色譜-串聯質譜(gas chromatography-tandem mass spectrometry,GC-MS/MS) 法[16-17]、液相色譜-串聯質譜法[18-19]等。其中,GC-MS/MS法具有靈敏度高、特異性強、分離效果好等優點,是多種農藥殘留定量分析的主要手段。為了保障豆乳產品的質量安全,避免受到農藥殘留污染的風險,本研究利用QuEChERS(quick, easy, cheap, effective, rugged, safe)的前處理方法,通過優化關鍵步驟,建立一種GC-MS/MS同時測定豆乳中12 種常見有機磷及氨基甲酸酯類農藥殘留量的方法。這將為其他植物乳中農藥殘留檢測提供參考。
1 材料與方法
1.1 材料與試劑
豆乳樣品:植選濃香豆乳、植選植物乳、植選濃香豆乳(黑芝麻、黑豆) 內蒙古伊利實業集團股份有限公司。
無水硫酸鎂(MgSO4)、氯化鈉、檸檬酸鈉、檸檬酸二鈉鹽倍半水合物(檸檬酸氫二鈉)、醋酸鈉(均為分析純) 美國安捷倫科技有限公司;乙酸乙酯(色譜純)、乙腈(農殘級)、乙二胺-N-丙基硅烷化硅膠(primary secondary amine,PSA)、十八烷基硅烷鍵合硅膠(C18)、石墨化炭黑、含12 種農藥(溴硫磷、三硫磷、毒蟲畏、毒死蜱、甲基毒死蜱、蠅毒磷、甲基嘧啶磷、仲丁威、苯硫威、異丙威、抗蚜威、殘殺威)的混標(100 μg/mL)、環氧七氯B(內標,1 mg/mL) 上海 安譜實驗科技股份有限公司。
1.2 儀器與設備
TQS9000氣相色譜-三重四極桿質譜聯用儀(配有電子轟擊離子源) 美國Thermo Fisher Scientific公司;Biofuge Stratos低溫高速離心機 德國Sigma公司;分析天平(感量0.000 1 g) 瑞士Mettler Toledo公司;TurboVap LV氮吹儀 瑞典Biotage公司;KS501渦旋振蕩器 德國IKA公司。
1.3 方法
1.3.1 樣品前處理
準確稱取10.0 g豆乳樣品于50 mL離心管中,加入10 mL乙腈,渦旋振蕩1 min;然后在冷水浴條件下加入4.0 g MgSO4、1.0 g氯化鈉、1.0 g檸檬酸鈉和0.5 g檸檬酸氫二鈉后,繼續振蕩5 min,4 ℃、4 200 r/min離心5 min;取7.0 mL上清液有機層至內含1 200 mg MgSO4、400 mg PSA和400 mg C18的15 mL離心管中,振蕩5 min,4 ℃、4 200 r/min離心5 min;取4.0 mL上清液至氮吹管中,40 ℃氮吹至近干;加入20 μL 5 μg/mL內標溶液,乙酸乙酯復溶至1.0 mL,過濾后供GC-MS/MS測定分析。
1.3.2 標準工作溶液配制
采用空白基質加標的方式,稱取不含待測農藥的樣品按上述操作制備樣品空白提取液,空白基質溶液氮氣吹干后,加入20 μL 5 μg/mL內標溶液,依次加入一定體積的混合標準品工作液,用乙酸乙酯稀釋定容至1.0 mL,配制成質量濃度梯度為5、10、20、50、100、150、200 ng/mL的12 種化合物的混合標準工作溶液。現用現配。
1.3.3 色譜條件
色譜柱:5%苯基-甲基聚硅氧烷石英毛細管色譜柱(30 m×0.25 mm,0.25 μm);升溫程序:40 ℃保持1.5 min,以25 ℃/min升溫至90 ℃,并保持1.5 min,以25 ℃/min升溫至180 ℃,以5 ℃/min升溫至280 ℃,以10 ℃/min升溫至300 ℃,保持5 min;進樣量1 μL;進樣方式:不分流進樣。
1.3.4 質譜條件
電離方式:電子轟擊源;電離能量70 eV;離子源溫度300 ℃;傳輸線溫度280 ℃;溶劑延遲6 min;監測方式:選擇反應監測模式,12 種農藥及環氧七氯B的保留時間、定量離子對、定性離子對和碰撞電壓參見表1。
1.3.5 結果計算
12 種農藥的含量計算公式如下。
式中:X為試樣中被測組分殘留量/(mg/kg);ρ為由標準工作曲線得到的被測組分質量濃度/(ng/mL);V為樣品溶液定容體積/mL;m為試樣質量/g;n為稀釋倍數;1 000為換算系數。
1.4 數據處理
使用Tracefinder 4.1軟件處理標準工作曲線,得到被測組分質量濃度;使用WPS Office 2020軟件繪制簇狀柱形圖。
2 結果與分析
2.1 前處理方法的確定
為了選擇合理的前處理方法,本研究參照植物源性食品基質,對GC法和GC-MS法測定有機磷和氨基甲酸酯類農藥殘留的國內標準進行總結。由表2可知,總體而言,前處理方法可分為液-液萃取、固相萃取柱及分散固相萃取3 種方式。其中,GB/T 5009.145—2003《植物性食品中有機磷和氨基甲酸酯類農藥多種殘留的測定》[11]的前處理步驟采用丙酮水溶液提取,二氯甲烷二次提取,硅膠柱凈化,最后濃縮定容。該方法的實驗步驟復雜,且需要消耗大量有機試劑,對環境及人員危害較大。NY/T 761—2008《蔬菜和水果中有機磷、有機氯、擬除蟲菊酯和氨基甲酸酯類農藥多殘留的測定》[20]為多個方法的合集,對于有機磷類農藥,采用液-液萃取,乙腈提取,過濾濃縮后定容;對于氨基甲酸酯農藥,采用乙腈提取,經氨基柱凈化,氮吹復溶上樣。該方法操作較麻煩,需要在80 ℃水浴條件下加熱濃縮,且樣品凈化不夠充分,不適用于復雜的樣品基質,不利于儀器長時間使用。對于DB34/T 1076—2009《蔬菜、水果、糧食、茶葉中40 種有機磷和氨基甲酸酯類農藥多殘留同時測定方法 氣相色譜法》[21]和GB 23200.8—2016《食品安全國家標準 水果和蔬菜中500 種農藥及相關化學品殘留量的測定 氣相色譜-質譜法》[22],前處理有了明顯改進,采用乙腈提取-固相萃取柱凈化的前處理方法,溶劑的使用量及操作步驟部分減少。GB 23200.113—2018《食品安全國家標準 植物源性食品中208 種農藥及其代謝物殘留量的測定 氣相色譜-質譜聯用法》[16]中除提供了乙腈提取-石墨化炭黑/氨基復合柱凈化的方法外,還推薦了QuEChERS前處理方法。研究表明,QuEChERS整個過程操作簡單、試劑用量少、環境污染小、處理時間短[23]。綜上所示,結合實際實驗操作,本研究最終確定采用QuEChERS前處理法。
2.2 萃取步驟中降溫方式的選擇
QuEChERS法提取時會加入MgSO4,可以更好地促使水相與有機相分層。然而MgSO4遇水會放出大量的熱,容易導致部分農藥分解,最終影響目標物的提取效率。為了避免這一問題的發生,本研究采用水傳導散熱的原理,快速釋放MgSO4遇水產生的熱量,降低實驗體系的溫度。優化后的實驗流程為在冷水浴條件下向離心管中加入鹽包。
將冷水浴降溫與傳統方式(常溫實驗環境)降溫進行對比實驗,每組做3 次平行。由圖1可知,2 種降溫方式的平均加標回收率分別為82.3%~97.6%和67.0%~85.8%;在冷水浴條件下,所有目標物的平均加標回收率優于傳統方式處理,其中異丙威效果最明顯,其平均加標回收率提升15.3%。因此,本研究選擇在冷水浴條件下進行實驗。
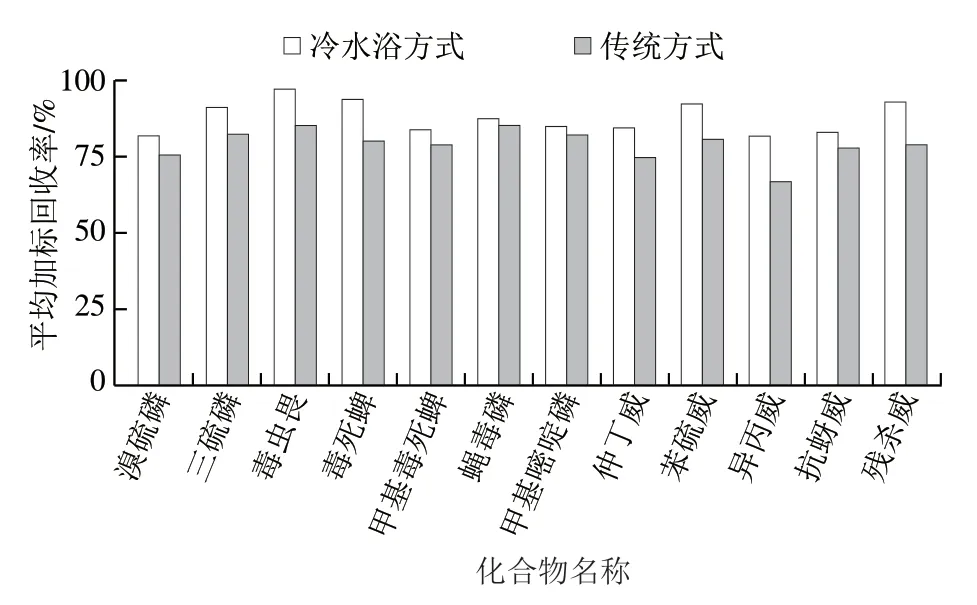
圖1 2 種降溫方式對12 種農藥平均加標回收率的影響 (加標量0.004 mg/kg)Fig. 1 Effect of two cooling methods on average recoveries of 12 pesticides at spiked concentration of 0.004 mg/kg
2.3 凈化步驟中凈化包的選擇
為了確定合適的QuEChERS凈化包,結合國標 GB 23200.113—2018《食品安全國家標準 植物源性食品中208 種農藥及其代謝物殘留量的測定 氣相色譜-質譜聯用法》中蔬菜、水果及谷物的前處理實驗,以及其他文獻調研的結果[16,24],本研究選取2 組通用型的凈化填料進行對比實驗:第1組為1 200 mg MgSO4、400 mg PSA和400 mg C18;第2組為885 mg MgSO4、150 mg PSA和15 mg GCB,每組做3 次平行。
由圖2可知,2 組實驗的平均加標回收率分別為87.7%~99.9%和76.4%~110.3%,除溴硫磷、蠅毒磷及殘殺威外,其余9 種目標物在MgSO4+PSA+C18條件下凈化,平均加標回收率更優。因此,本研究選擇通用型凈化包1 200 mg MgSO4、400 mg PSA和400 mg C18。
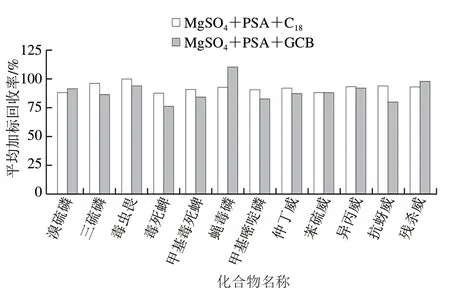
圖2 2 種凈化包對12 種農藥平均加標回收率的影響 (加標量0.004 mg/kg)Fig. 2 Effect of two dispersive solid phase extraction kits on average recoveries of 12 pesticides at spiked concentration of 0.004 mg/kg
2.4 標準曲線和定量限
本研究采用內標法進行定量,將基質標準溶液依次進樣后采集數據,以目標物和內標的定量離子對峰面積比值為縱坐標,二者之間的質量濃度比為橫坐標,繪制標準曲線。
由表3可知,在5~200 ng/mL質量濃度范圍內,12 種有機磷及氨基甲酸酯類農藥的標準曲線線性關系良好,相關系數均大于0.99。12 種農藥的定量限均為 0.004 mg/kg,滿足信噪比(RS/N)≥10的要求。

表3 12 種農藥的標準曲線和定量限Table3 Calibration curves and LOQs for 12 pesticides
2.5 方法回收率和精密度
選擇空白豆乳樣品進行0.004、0.008、0.040 mg/kg 3 個不同水平的加標實驗,每個水平均做6 次平行,測定加標回收率及相對標準偏差(relative standard deviation,RSD)。
由表4可知,12 種農藥的加標回收率為71.8%~114.9%,RSD為4.6%~13.3%,說明該方法滿足要求,適用于豆乳中目標農藥殘留的測定。在實際樣品檢測中,利用所建立的方法對市售的5 批次豆乳進行12 種有機磷和氨基甲酸酯類農藥的檢測,結果均未檢出。

表4 12 種農藥的加標回收率及RSD(n= 6)Table4 Recoveries and RSDs for 12 pesticides (n = 6)
2.6 實際樣品測定
隨機抽取3 種常見豆乳產品進行檢測,分別稱取10 g植選濃香豆乳、植選植物乳、植選濃香豆乳(黑芝麻、黑豆),按照方法操作步驟進行12 種農藥殘留檢測,結果均未檢出,與樣品實際情況相符,表明此方法適用。
3 結 論
本研究提供了一種同時測定豆乳中12 種有機磷及氨基甲酸酯類農藥殘留的GC-MS/MS方法。在其他食品基質文獻的基礎上,采用QuEChERS前處理方法,在提取步驟中引入冷水浴降溫方式,較好地提升了目標物的加標回收率;在凈化步驟中對通用型凈化包進行了加標回收率對比實驗,確定了合適比例的凈化包;同時采用內標法進行定量。方法學驗證實驗結果顯示:12 種農藥的定量限均為0.004 mg/kg,3 個不同添加水平下的加標回收率為71.8%~114.9%,RSD為4.6%~13.3%,加標回收率和精密度滿足要求。該方法具有快速、高效、準確的優點,適用于豆乳中有機磷及氨基甲酸酯農藥殘留的測定,也可為其他植物乳中農藥殘留檢測方法的研究及應用提供參考。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創版(2016年10期)2016-11-25 10:28:30
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
