通宣理肺丸HPLC指紋圖譜建立及7種成分測定
2021-10-26 12:56:10張國強邱國玉石曉峰
中成藥 2021年10期
李 運, 王 苗, 張國強, 邱國玉, 石曉峰, 戴 忠
(1.蘭州市食品藥品檢驗檢測研究院/甘肅省種植中藥材外源性污染物監測工程研究中心,甘肅 蘭州 730000;2.蘭州大學藥學院,甘肅 蘭州 730000;3.甘肅中醫藥大學藥學院,甘肅 蘭州 730000;4.甘肅省醫學科學研究院,甘肅 蘭州 730050;5.中國食品藥品檢定研究院,北京 100050)
通宣理肺丸由紫蘇葉、前胡、桔梗等11味藥材組成[1],方中君藥紫蘇葉、麻黃辛溫宣肺,發散風寒;臣藥前胡、桔梗輔助君藥宣通肺氣,杏仁潤肺止咳,半夏、陳皮、茯苓燥濕化痰;佐以黃芩清肺以防肺氣郁久化熱,枳殼行氣消痰;甘草為使藥,調和諸藥,全方共奏通宣理肺之功效,臨床應用較廣,但現行質量標準不能較全面的對其質量進行控制。因此,需借鑒指紋圖譜[2]、特征圖譜[3]、多成分測定[4-5]、數理統計[6]等方法,充分體現中藥整體或部分化學特性的譜圖,最大限度地表征藥效相關成分或特征成分[7],從而在中藥質量控制與評價中發揮重要的作用。
近年來,有學者對通宣理肺丸中橙皮苷、柚皮苷[8]、黃芩苷[9]、鹽酸麻黃堿[10]、白花前胡甲素、白花前胡乙素[11]含量進行了測定;姚蓉等[12]篩查了前胡投料情況,并對其特征成分含量進行測定;孫國祥等[13]采用毛細管區帶電泳,建立全方指紋圖譜。本實驗在前期報道及劉昌孝院士[14-15]提出“中藥質量標志物”概念的基礎上,選擇與中藥質量特性密切相關的標志物,建立通宣理肺丸HPLC指紋圖譜,并對甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素含量進行測定,以期為該制劑質量控制提供參考。
1 材料
1.1 儀器 Waters e2695液相色譜儀(美國Waters公司);SBL-10DT超聲波恒溫清洗機(寧波新芝生物科技股份有限公司);XSE205DU電子天平(瑞士梅特勒-托利多公司);Milli-Q IQ7000超純水機(美國默克密理博公司);ChemPattern化學計量學軟件[科邁恩(北京)科技有限公司]。
1.2 試劑與藥物 甘草苷(批號111610-201908,純度95%)、柚皮苷(批號110722-201815,純度91.7%)、橙皮苷(批號110721-202019,純度95.3%)、新橙皮苷(批號111857-201804,純度99.4%)、甘草酸銨(批號110731-201619,純度96.2%)對照品均購自中國食品藥品檢定研究院;黃芩苷(批號8369,純度96.2%)、白花前胡甲素(批號5636,純度99.2%)對照品均購自上海詩丹德生物技術有限公司。通宣理肺丸(大蜜丸)31批,編號S1~S31,具體見表1。乙腈、甲醇為色譜純;其他試劑為分析純;純化水為實驗室自制。
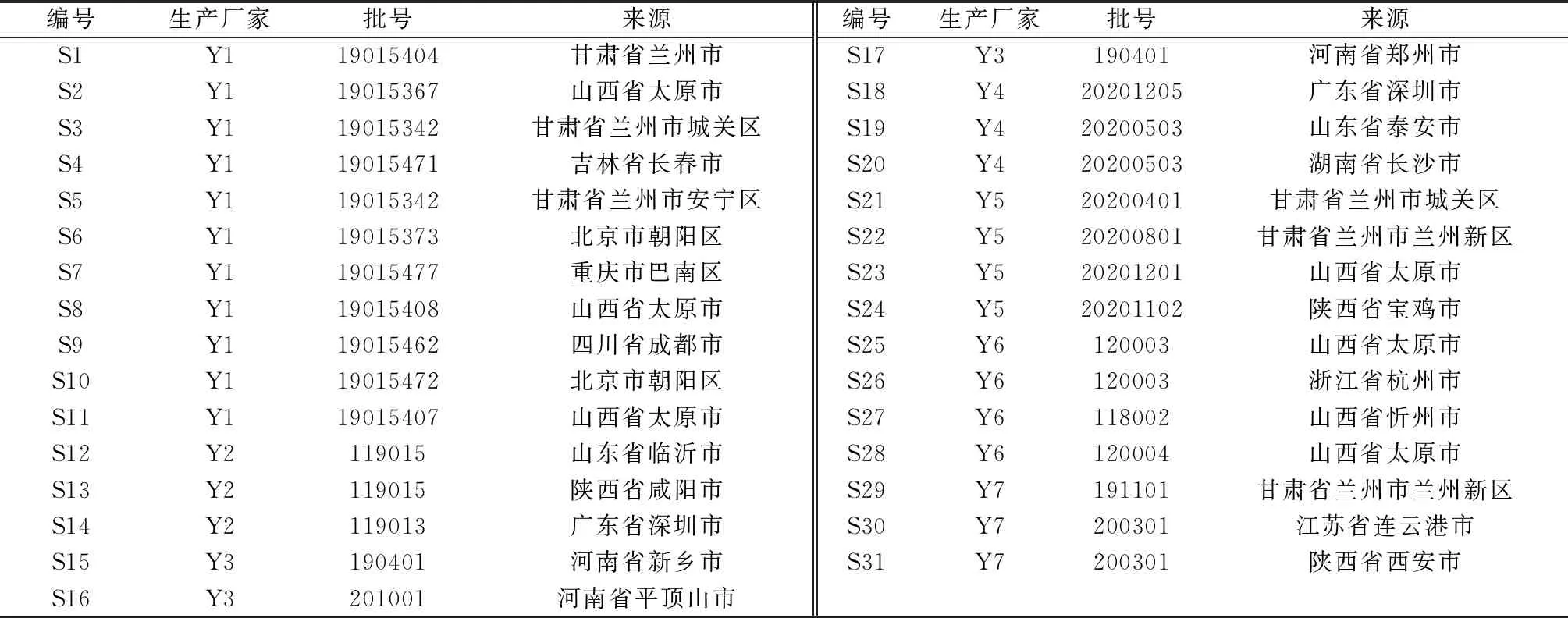
表1 樣品信息
2 方法與結果
2.1 色譜條件 CAPCELL PAK C18MG色譜柱(250 mm×4.6 mm,5 μm);流動相水(含0.1%甲酸)(A)-80%乙腈(B),梯度洗脫(0~5 min,15%B;5~10 min,15%~20%B;10~20 min,20%~25%B;20~30 min,25%B;30~40 min,25%~48%B;40~45 min,48%~70%B;45~55 min,70%~90%B;56~60 min,90%~15%B);體積流量1.0 mL/min;柱溫30 ℃;檢測波長250 nm;進樣量10 μL。
2.2 溶液制備
2.2.1 對照品溶液 精密稱取各對照品適量,置于10 mL量瓶中,甲醇溶解并定容,即得(甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素質量濃度分別為1.003 2、0.989 4、0.582 3、1.064 6、0.575 3、1.335 3、1.174 5 mg/mL),4 ℃下保存備用。
2.2.2 供試品溶液 取樣品(S3)適量,剪碎,混勻,取約1 g,精密稱定,加硅藻土1 g,研勻,置于150 mL錐形瓶中,精密加入70%乙醇25.00 mL,稱定質量,超聲提取30 min,靜置冷卻,70%乙醇補足減失的質量,0.45 μm微孔濾膜過濾,取續濾液,即得。
2.2.3 陰性樣品溶液 按處方比例及工藝,分別制得缺甘草,缺陳皮,缺陳皮、枳殼,缺黃芩,缺前胡,缺甘草、黃芩、前胡、陳皮、枳殼的陰性樣品,按“2.2.2”項下方法制備,即得。
2.3 HPLC指紋圖譜建立
2.3.1 精密度試驗 取樣品(S3),按“2.2.2”項下方法制備供試品溶液,在 “2.1”項下色譜條件進樣測定6次,以18號峰(黃芩苷)為參照,測得各共有峰相對保留時間 RSD為0.052%~0.501%,相對峰面積RSD為0.856%~2.650%,表明儀器精密度良好。
2.3.2 重復性試驗 取樣品(S3),按“2.2.2”項下方法平行制備6份供試品溶液,在“2.1”項下色譜條件進樣測定,以18號峰(黃芩苷)為參照,測得各共有峰相對保留時間RSD為0.026%~1.000%,相對峰面積RSD為1.017%~2.498%,表明該方法重復性良好。
2.3.3 穩定性試驗 取樣品(S3),按“2.2.2”項下方法制備供試品溶液,于0、2、4、8、12、24 h在“2.1”項色譜條件下進樣測定,以18號峰(黃芩苷)為參照,測得各共有峰相對保留時間RSD為0.035%~1.638%,相對峰面積RSD為1.005%~2.826%,表明溶液在24 h內穩定性良好。
2.3.4 圖譜生成 取31批樣品(S1~S31),按“2.2.2”項下方法制備供試品溶液,在“2.1”項下色譜條件進樣測定,結果見圖1,將所得數據導入ChemPattern軟件,設置積分條件(斜率0.01,最小相對峰高0.1%,最小百分比峰面積0.25%,積分開始3,積分結束300),以75%(高四分位數)為共有峰篩選條件,采用高斯曲線模擬生成共有模式,發現39個共有峰,見圖2。
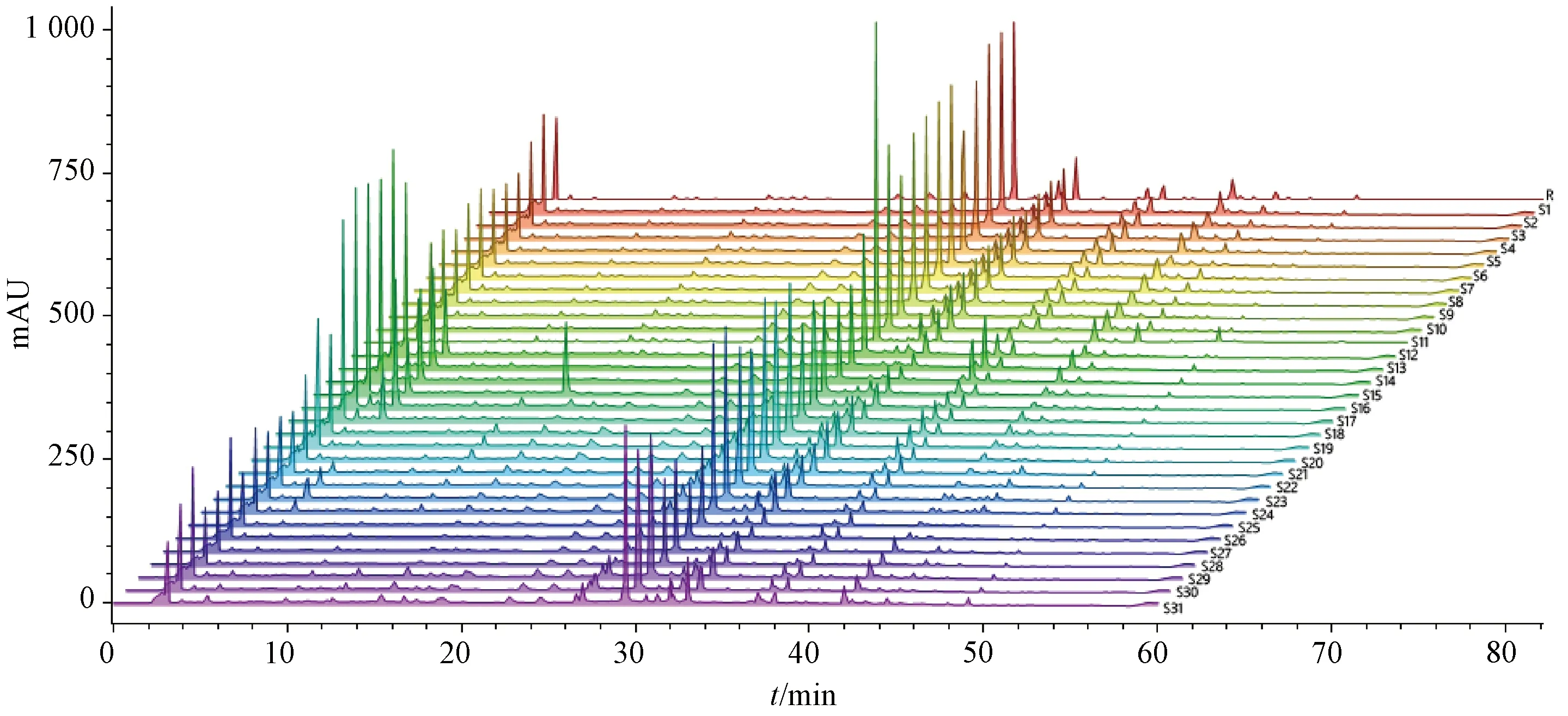
圖1 31批樣品HPLC指紋圖譜
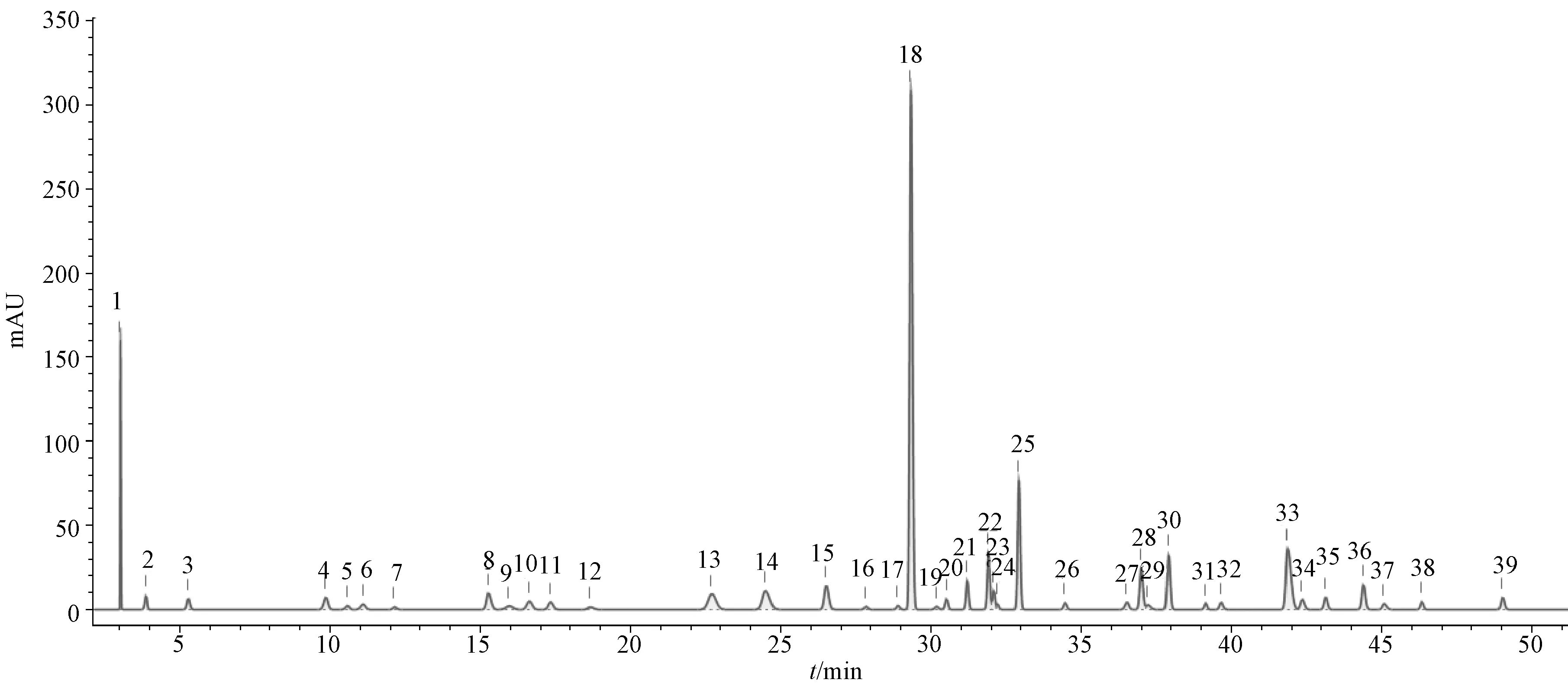
10.甘草苷 13.柚皮苷 14.橙皮苷 15.新橙皮苷 18.黃芩苷 30.甘草酸 39.白花前胡甲素 10.liquiritin 13.naringin 14.hesperidin 15. neo-hesperidin 18.baicalin 30. glycyrrhizic acid 39.praeruptorin A
2.3.5 相似度分析 采用ChemPattern軟件,以HPLC指紋圖譜共有模式為參照圖譜,通過夾角余弦法計算各樣品相似度,見表2,可知相似度為0.16~0.99,其中有77.4%大于0.95,表明不同企業樣品質量差異較大,其中Y2、Y3企業有3批低于0.6,均存在部分特征峰丟失或者峰面積偏小的情況。
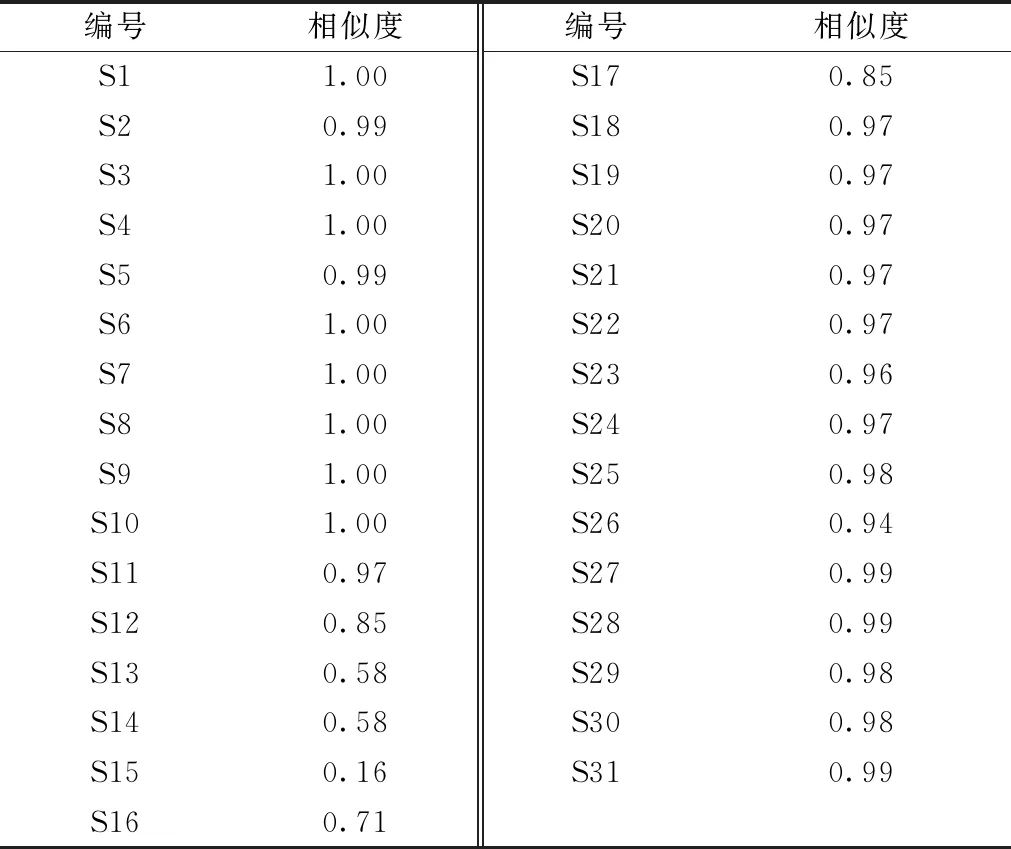
表2 31批樣品相似度
2.3.6 聚類分析 采用ChemPattern軟件將31批樣品HPLC指紋圖譜數據進行標準化后,以街區距離為度量,通過遠鄰法進行聚類分析,結果見圖3。
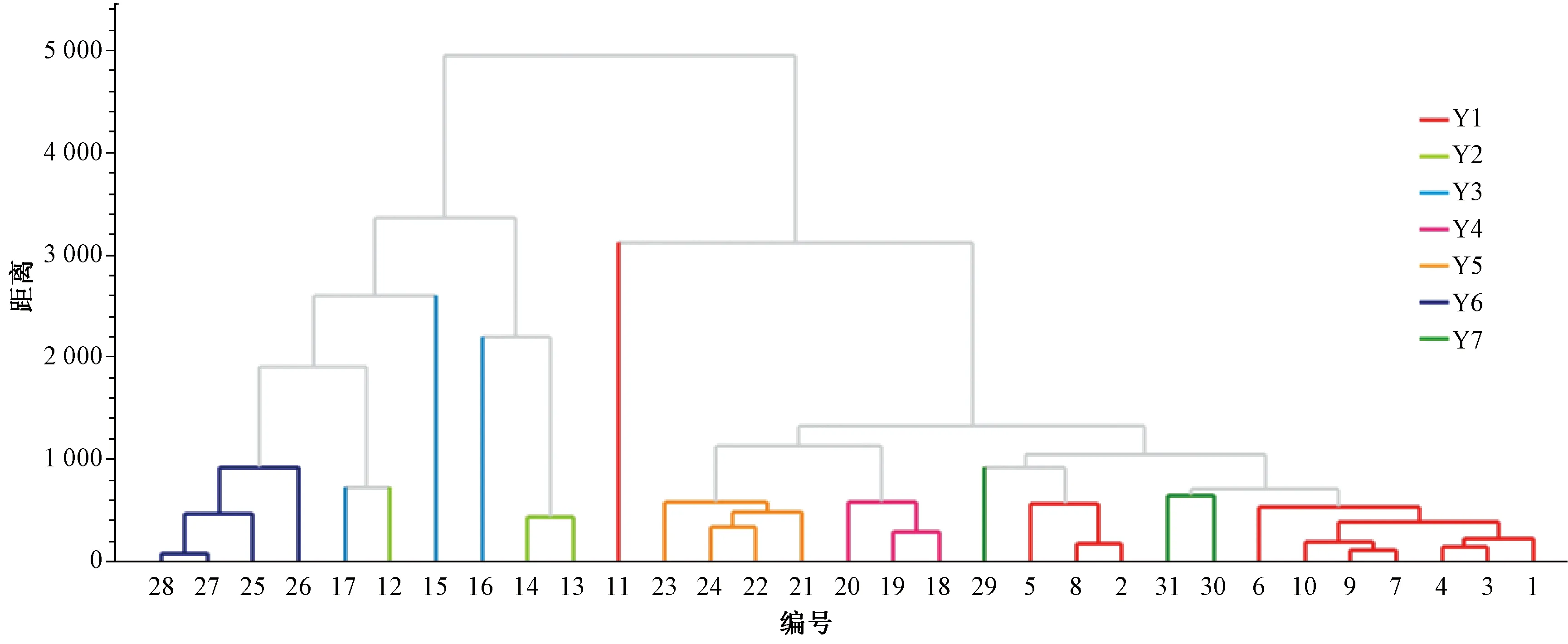
圖3 31批樣品聚類分析圖
2.3.7 主成分分析 采用ChemPattern軟件進行主成分分析,結果見圖4。由此可知,第一主成分(PC1)、第二主成分(PC2)、第三主成分(PC3)貢獻率分別為65.13%、25.64%、3.99%,總計94.76%,能較好地反映各批樣品之間的差異;峰18、3、28、1、14、33、12、25、39是主要標志性成分,與對照品圖譜比對,可知峰14為橙皮苷,峰18為黃芩苷,峰39為白花前胡甲素。
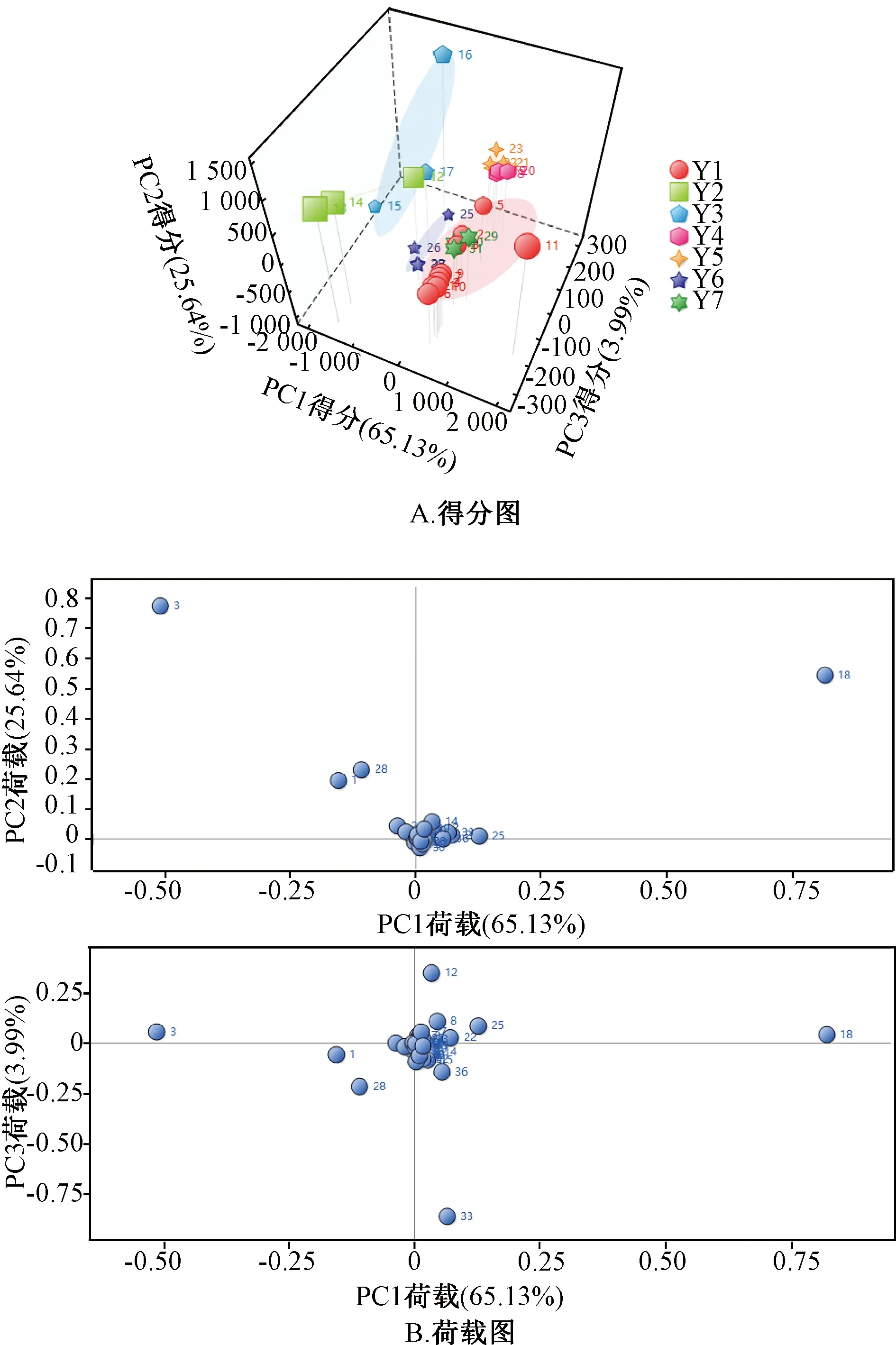
圖4 31批樣品主成分分析圖
2.4 含量測定
2.4.1 專屬性試驗 取“2.2”項下對照品、供試品、陰性樣品溶液,在“2.1”項下色譜條件進樣測定,結果見圖5。由此可知,各成分分離度良好,丸劑中其他共存物質對測定結果無干擾,理論塔板數以黃芩苷(5號峰)計,不得低于6 000。
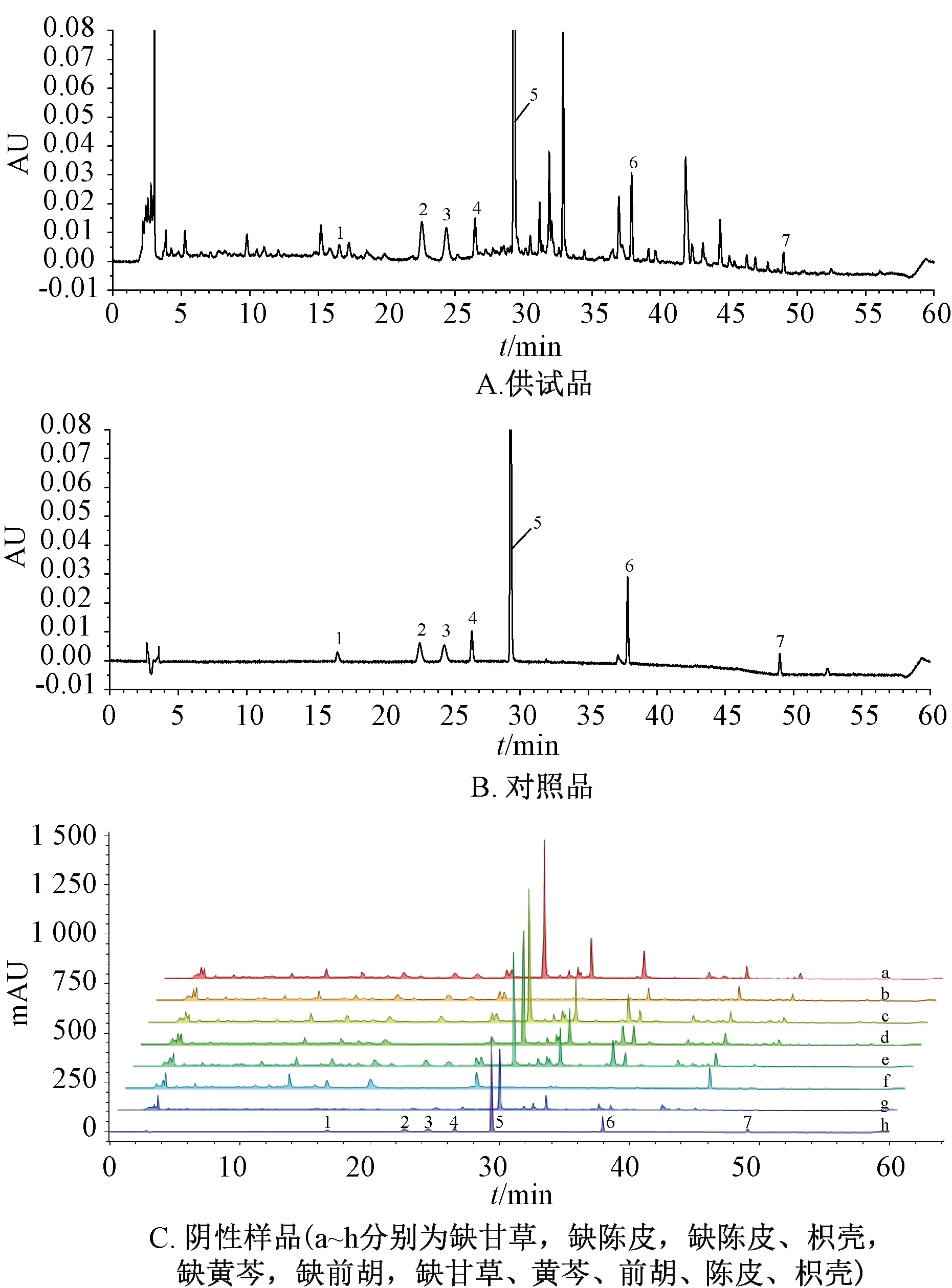
1.甘草苷 2.柚皮苷 3.橙皮苷 4.新橙皮苷 5.黃芩苷 6.甘草酸 7.白花前胡甲素
2.4.2 線性關系考察 精密吸取“2.2.1”項下對照品溶液適量,置于10 mL量瓶中,甲醇定容至刻度,搖勻,在“2.1”項色譜條件下進樣測定。以對照品質量濃度為橫坐標(X),峰面積為縱坐標(Y)進行回歸,結果見表3,可知各成分在各自范圍內線性關系良好。

表3 各成分線性關系
2.4.3 精密度試驗 取“2.3.2”項下對照品溶液(甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素質量濃度分別為100.32、98.94、58.23、106.46、57.53、133.53、117.45 μg/mL),按“2.2.2”項下方法制備供試品溶液,在“2.1”項色譜條件下進樣測定6次,測得甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素峰面積RSD分別為2.8%、2.3%、1.7%、0.6%、0.4%、0.8%、1.7%,表明儀器精密度良好。
2.4.4 重復性試驗 取同一批樣品(S3),按“2.2.2”項下方法平行制備6份供試品溶液,在“2.1”項色譜條件下別進樣測定,測得甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素含量RSD分別為1.88%、2.15%、2.02%、1.93%、1.78%、1.45%、2.19%,表明該方法重復性良好。
2.4.5 穩定性試驗 取同一批樣品(S3),按“2.2.2”項下方法制備供試品溶液,于0、2、4、8、12、24 h在“2.1”項色譜條件下進樣測定,測得甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素峰面積RSD分別為2.49%、2.32%、2.00%、2.12%、1.05%、1.24%、2.08%,表明溶液在24 h內穩定性良好。
2.4.6 加樣回收率實驗 精密稱取各成分含量已知的樣品(S3)0.5 g,共6份,加入對照品溶液,按“2.2.2”項下方法制備供試品溶液,在“2.1”項色譜條件下進樣測定,計算回收率。結果,甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素平均加樣回收率分別為99.89%、113.3%、101.2%、105.2%、93.10%、98.10%、106.6%,RSD分別為2.29%、2.58%、2.52%、1.61%、3.86%、1.25%、2.41%。
2.5 樣品含量測定 取31批樣品,每批2份,按“2.2.2”項下方法制備供試品溶液,在“2.1”項色譜條件下進樣測定,外標法計算含量,結果見表4。
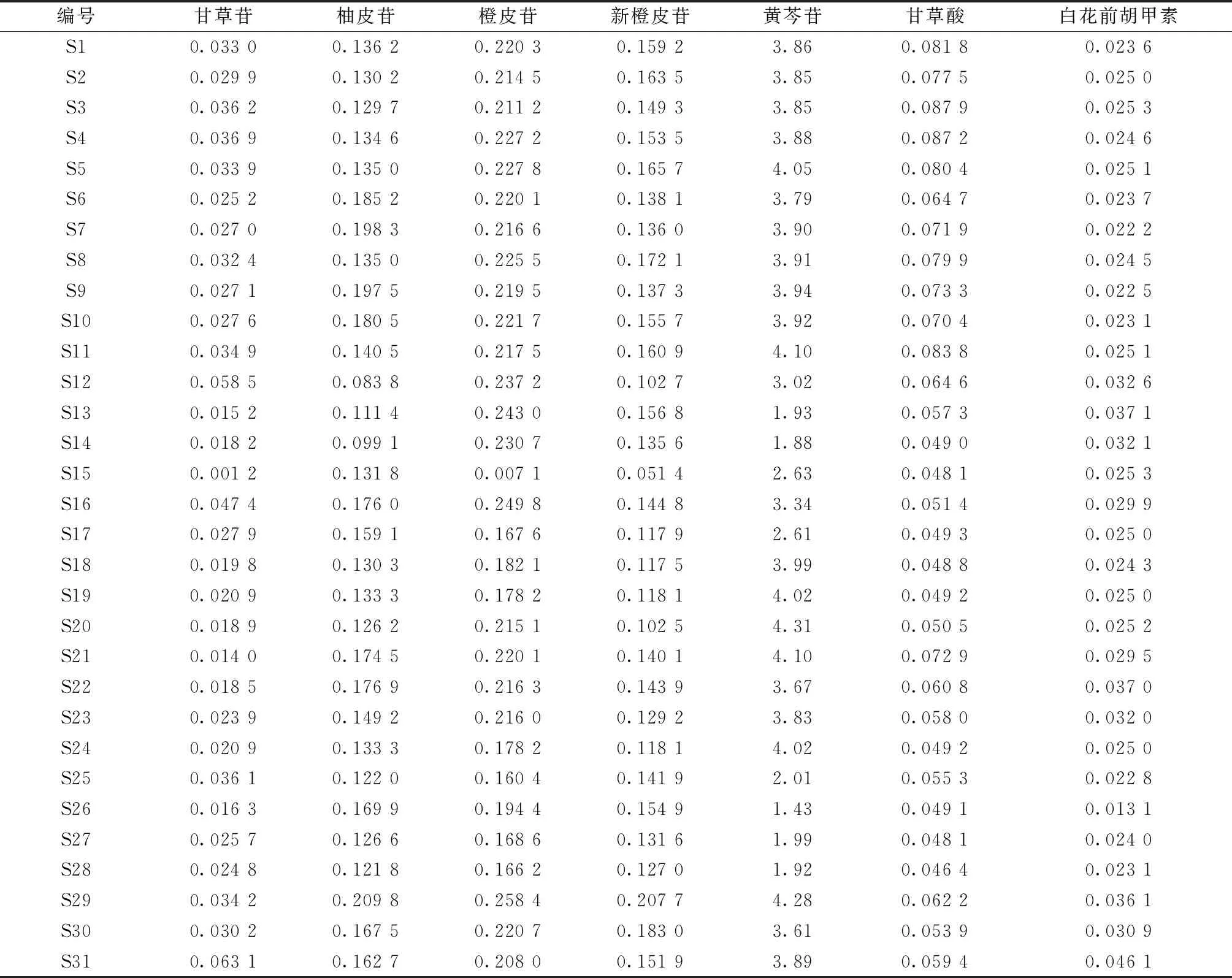
表4 各成分含量測定結果(%,n=3)
3 討論
3.1 色譜條件優化
3.1.1 流動相 本實驗考察了90%乙腈-水(含0.2%磷酸)、80%乙腈-水(含0.2%磷酸)、80%乙腈-水(含0.1%甲酸),發現80%乙腈-水(含0.1%甲酸)梯度洗脫60 min時,各成分色譜峰峰形較好。
3.1.2 檢測波長 本實驗采用二極管陣列檢測器對各成分進行全波長掃描,綜合考慮不同吸收波長處的圖譜響應強度,最終確定250 nm作為檢測波長,此時各成分檢測靈敏度較好,干擾小。
3.2 提取條件優化 本實驗考察了不同提取方法(超聲、回流)、提取溶劑(70%乙醇、70%甲醇、甲醇)、提取時間(30 min、1 h、1.5 h),結合綠色分析的需要,最終確定25 mL 70%乙醇超聲提取30 min作為提取條件。
3.3 含量測定分析 企業Y1、Y4、Y5、Y6、Y7樣品之間的相似度均大于0.95,質量均一性良好;Y2、Y3不同批次及同一批次樣品之間存在較大差異。31批樣品中,甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素質量分數范圍分別為0.001 2%~0.063 1%、0.083 8%~0.209 8%、0.007 1%~0.258 4%、0.051 4%~0.207 7%、1.43%~4.31%、0.046 4%~0.087 9%、0.013 1%~0.046 1%,差異較明顯,故對通宣理肺丸質量的整體控制方法有待進一步完善。
4 結論
本實驗建立了通宣理肺丸HPLC指紋圖譜,并同時測定了甘草苷、柚皮苷、橙皮苷、新橙皮苷、黃芩苷、甘草酸、白花前胡甲素含量,發現指紋圖譜結合數理統計能很好地從整體上表征該制劑特性,而且該方法簡便準確,重復性、穩定性良好,可為其質量控制提供參考。
