磁性MOFs材料Fe3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附性能
2021-10-09 09:58:40古建杉劉德蓉袁果園
原子能科學技術(shù) 2021年10期
關(guān)鍵詞:模型
古建杉,劉德蓉,周 倩,李 建,湯 毅,袁果園,*,熊 偉,劉 寧
(1.重慶科技學院 化學化工學院,重慶 401331; 2.四川大學 原子核科學技術(shù)研究所 輻射物理及技術(shù)教育部重點實驗室, 四川 成都 610064)
60Co是一種堆造放射性同位素,由慢中子源轟擊59Co獲得,半衰期為5.27 a。因其能釋放γ射線,廣泛應(yīng)用于輻射育種、蟲害防治、輻射消毒和放射治療等領(lǐng)域[1-2]。在60Co的生產(chǎn)和使用過程中,不可避免地會產(chǎn)生含60Co放射性廢水,這些廢水未經(jīng)處理直接排放,極易對環(huán)境和人體造成放射性污染和輻射損傷。因此,從含60Co放射性廢水中分離60Co對環(huán)境治理具有十分重要的意義。
金屬-有機骨架材料(MOFs)是由無機單元(金屬離子中心或金屬簇等)和有機連接劑(羧酸鹽、偶氮鹽等)橋連而成的多空配位聚合物,具有功能性強、孔隙率高、比表面積大等特點,廣泛用于金屬離子的分離研究[3]。Huang等[4]采用柔性配位合成后修飾策略制備了鉍硫醇功能化HKUST-1材料并用于Hg2+的分離,吸附量為264 mg/g。Ahmed等[5]將乙二胺嫁接在具有游離羧基的UiO-66上得到UiO-66-COOH-ED,其對Gd3+的最大吸附量達78.4 mg/g。Yuan等[6]將希夫堿接枝到鋯基MOFs材料UiO-66-NH2上制得希夫堿功能化的MOFs材料UiO-66-Schiff base,對Co(Ⅱ)的最大吸附量達256.4 mg/g。隨后,他通過結(jié)合密度泛函理論(DFT)計算和合成后修飾法進一步研究了不同功能基團(酯基、酰胺基、腈基、磺酸基)修飾的MOFs材料對Co(Ⅱ)的分離性能,結(jié)果發(fā)現(xiàn)幾種基團功能化的MOFs材料對Co(Ⅱ)均具有較好的吸附效果且與理論計算一致。然而,通過合成后修飾盡管能提高MOFs材料對Co(Ⅱ)的吸附量,卻存在制備工藝復(fù)雜、接枝量難以準確計算等問題。因此,本工作希望通過一步法將磺酸基功能化的鋯基MOFs材料UiO-66-SO3H與SiO2包裹的磁性顆粒Fe3O4相結(jié)合制得功能化的磁性MOFs材料Fe3O4@SiO2@UiO-66-SO3H,并用于Co(Ⅱ)的分離研究,以解決合成后修飾制備工藝繁瑣等問題,實現(xiàn)Co(Ⅱ)的磁性高效分離。
1 實驗
1.1 主要試劑與儀器
氫氧化鈉,分析純,成都科隆有限公司;N,N-二甲基甲酰胺、對苯二甲酸、2-磺酸基對苯二甲酸單鈉、四氯化鋯、2-(環(huán)己基氨基)-乙磺酸(CHES)、Fe3O4、六水硝酸鈷,分析純,中國麥克林有限公司。
DHG-9023A電熱鼓風干燥箱,合恒公司;T6紫外可見分光光度計,普析通用公司;THZ-82AS冷凍水浴恒溫振蕩器,榮華公司;BRUKER TENSOR Ⅱ傅立葉紅外光譜儀(FT-IR),德國布魯克公司;JSM-7001F熱場發(fā)射掃描電子顯微鏡,日本電子公司;XRD-7000 X射線衍射儀(XRD)、AXIS-ULTRA DLD型X射線光電子能譜儀(XPS),日本島津公司;EZ7振動樣品磁強計,美國MicroSense公司。
1.2 實驗水樣
稱取4.938 0 g 六水硝酸鈷于250 mL燒杯中,加入含2%硝酸的去離子水溶解后轉(zhuǎn)移至1 000 mL容量瓶中稀釋定容,即得到1 000 mg/L Co(Ⅱ)儲備液。實驗前用0.050 0 mol/L的2-環(huán)己胺基乙磺酸(CHES)溶液(緩沖溶液)稀釋至相應(yīng)濃度,再用氫氧化鈉溶液或硝酸溶液進行pH值調(diào)節(jié)。
1.3 Fe3O4@SiO2@UiO-66-SO3H制備
1) Fe3O4@SiO2制備
將1.50 g Fe3O4加入到7.5 mL 氨水和240 mL無水乙醇混合溶液中,機械攪拌下滴加9 mL正硅酸乙酯和30 mL無水乙醇的混合溶液,常溫下攪拌反應(yīng)12 h,所得產(chǎn)品使用磁鐵分離并用超純水和無水乙醇洗滌數(shù)次,在60 ℃下真空干燥即得Fe3O4@SiO2。
2) Fe3O4@SiO2@UiO-66-SO3H制備
取2.38 g ZrCl4、1.34 g對苯二酸、0.540 g 2-磺酸基對苯二甲酸單鈉溶于180 mL DMF和20 mL冰乙酸中,再加入1.20 g Fe3O4@SiO2,超聲15 min后轉(zhuǎn)入500 mL圓底燒瓶中,120 ℃油浴機械攪拌24 h,待溶液冷卻至室溫后,產(chǎn)品用磁鐵進行分離,并用蒸餾水和DMF洗滌數(shù)次,所得樣品在80 ℃下真空干燥即得Fe3O4@SiO2@UiO-66-SO3H。
1.4 Fe3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附
稱取10 mg 吸附劑于250 mL錐形瓶中,加入100 mL調(diào)節(jié)pH值后的一定濃度的Co(Ⅱ)溶液,放入恒溫振蕩箱中,在一定溫度下進行振蕩吸附。吸附完成后,使用磁鐵對吸附劑進行磁性分離,取上層清液并用紫外分光光度計測定其Co(Ⅱ)濃度。采用下式計算吸附量:
(1)
其中:qt為t時刻的吸附量,mg/g;c0和ct分別為溶液中Co(Ⅱ)的初始濃度和t時刻的濃度,mg/L;V為溶液體積,mL;m為吸附劑質(zhì)量,g。
2 結(jié)果與討論
2.1 吸附劑的表征
Fe3O4@SiO2和Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)前后的XRD譜示于圖1a。從圖1a可知,磁基體Fe3O4@SiO2(2θ=30.1°、35.5°、43.1°、57.1°)及UiO-66-SO3H包裹后的磁性Fe3O4@SiO2@UiO-66-SO3H材料(2θ=7.3°、8.4°、17.0°、25.7°)的XRD譜與文獻報道的Fe3O4[7]和UiO-66[8]的XRD譜一致,表明成功制得Fe3O4@SiO2和Fe3O4@SiO2@UiO-66-SO3H。

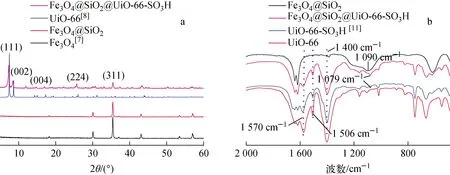
圖1 磁性MOFs材料的XRD譜(a)和FT-IR譜(b)Fig.1 XRD (a) and FT-IR (b) patterns of magnetic MOFs
Fe3O4@SiO2@UiO-66-SO3H的SEM圖像示于圖2,其N2吸附-脫附曲線示于圖3。從圖2可見,材料呈堆積狀態(tài),表面具有分布均勻的細小塊狀體,塊狀體棱角分明且表面較為光滑。根據(jù)圖3可得到材料的孔結(jié)構(gòu)參數(shù),與磁基體Fe3O4@SiO2相比,吸附劑的比表面積從29.4 m2/g增加至739.6 m2/g,孔容從0.023 cm3/g增加至0.047 cm3/g,而平均孔徑從7.12 nm降低至3.56 nm,進一步證明所制備吸附劑為Fe3O4@SiO2@UiO-66-SO3H,且該吸附劑兼具磁基體的磁性和MOFs材料的多孔結(jié)構(gòu)等優(yōu)異性能。

圖2 Fe3O4@SiO2@UiO-66-SO3H的SEM圖像Fig.2 SEM image of Fe3O4@SiO2@UiO-66-SO3H

圖3 Fe3O4@SiO2和Fe3O4@SiO2@UiO-66-SO3H 的N2吸附-脫附等溫線Fig.3 N2 adsorption-desorption isotherm for Fe3O4@SiO2 and Fe3O4@SiO2@UiO-66-SO3H
Fe3O4@SiO2和Fe3O4@SiO2@UiO-66-SO3H的磁滯回歸曲線示于圖4。從圖4可看出,此材料的飽和磁化強度為12.4 A·m2/kg與磁基體Fe3O4@SiO2的飽和磁化強度(48.4 A·m2/kg)相比有所下降,這是由于磁基體表面包裹了一層非磁性的UiO-66-SO3H所致。

圖4 Fe3O4@SiO2@UiO-66-SO3H的磁滯回歸曲線Fig.4 Hysteresis regression curve of Fe3O4@SiO2@UiO-66-SO3H
2.2 Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)的影響因素
1) 初始pH值
c0=10 mg/L、溫度298 K、t=24 h、m=10 mg下,初始pH值對Fe3O4@SiO2和Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)的影響示于圖5。由圖5可見,在實驗所設(shè)初始pH值下,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附量均較Fe3O4@SiO2的高。表明UiO-66-SO3H功能化后的材料吸附性能更好。當初始pH值由4增加到7時,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附量隨溶液初始pH值的增加而增加,但總的吸附量較低,這主要是由于pH≤7時,溶液中存在大量的質(zhì)子與Fe3O4@SiO2@UiO-66-SO3H的吸附位點的結(jié)合較與Co(Ⅱ)的結(jié)合更強,因此Co(Ⅱ)的吸附量較低[12]。當初始pH值由7增加到9時,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)吸附量顯著增加,這是因為隨著初始pH值的增加,溶液中質(zhì)子逐漸減少,F(xiàn)e3O4@SiO2@UiO-66-SO3H暴露出更多的吸附位點與Co(Ⅱ)結(jié)合,吸附量逐漸增加。Fe3O4@SiO2@UiO-66-SO3H在初始pH=9時的吸附量較高,主要是由于此時已有部分Co(Ⅱ)以Co(OH)2形式沉淀[13]。因此,后續(xù)研究選擇在初始pH=8.3下進行。
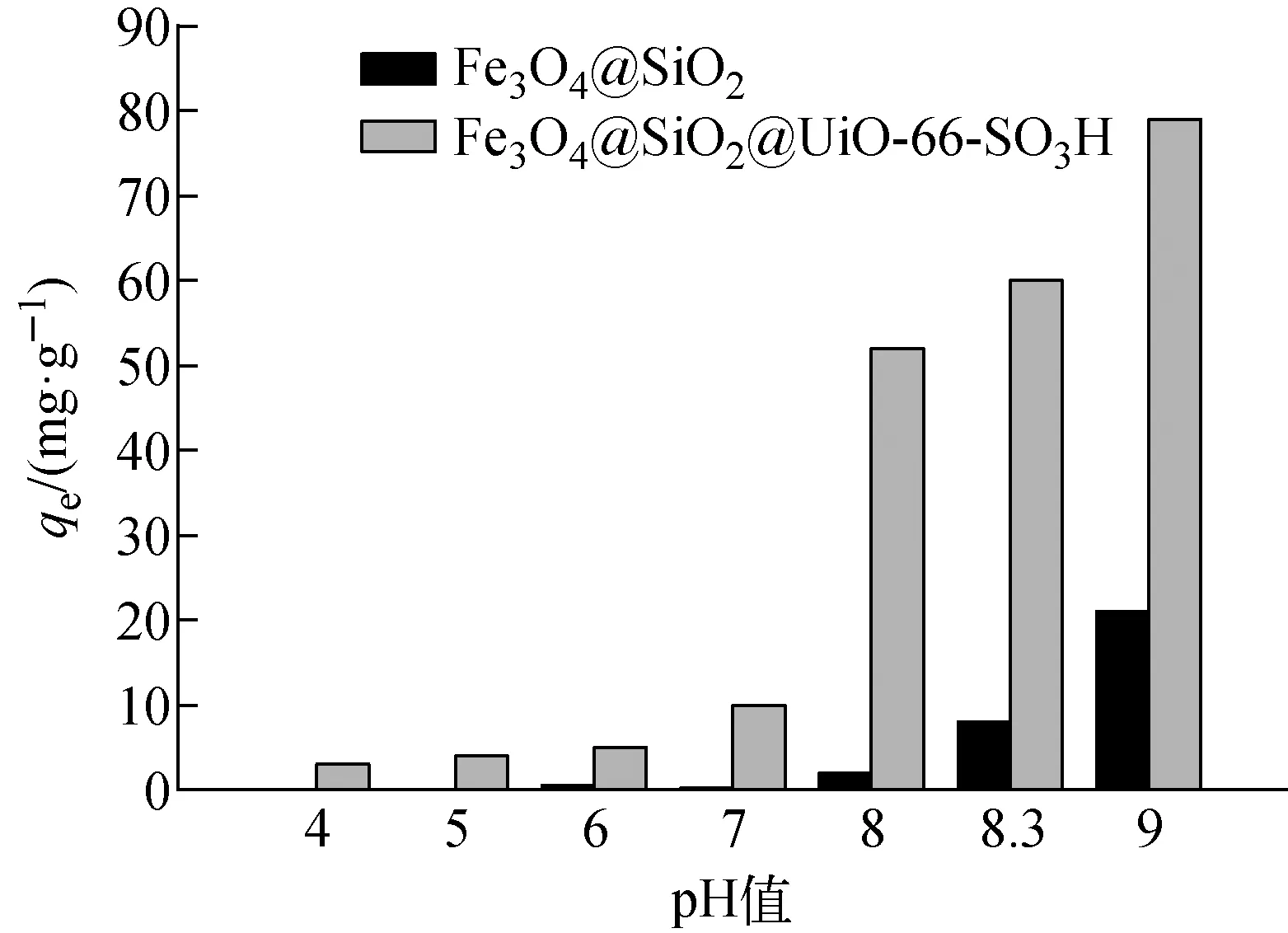
圖5 初始pH值對Fe3O4@SiO2和 Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)的影響Fig.5 Effect of initial pH on adsorption of Co(Ⅱ) on Fe3O4@SiO2 and Fe3O4@SiO2@UiO-66-SO3H
2) 吸附時間
c0=10 mg/L、溫度298 K、初始pH=8.3、m=10 mg下,吸附時間對Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)的影響示于圖6。由圖6可見,在前4 h內(nèi)Fe3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附是一快速過程,此后隨著時間的延長,吸附速率逐漸降低并趨于平衡,最大吸附量為62 mg/g,吸附平衡時間為24 h。

圖6 吸附時間對Fe3O4@SiO2@UiO-66-SO3H 吸附Co(Ⅱ)的影響Fig.6 Effect of time on adsorption of Co(Ⅱ) on Fe3O4@SiO2@UiO-66-SO3H
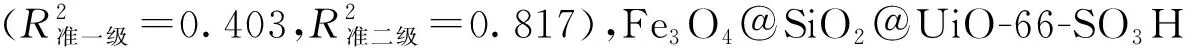
ln(qe-qt)=lnqe-k1t
(2)
(3)
式中:qe為平衡吸附量,mg/g;k1(min-1)和k2(g/(mg·min))分別為準一級動力學模型和準二級動力學模型的吸附速率常數(shù)。
3) Co(Ⅱ)初始濃度
t=24 h、溫度298 K、初始pH=8.3、m=10 mg下,Co(Ⅱ)初始濃度對Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)的影響示于圖7。由圖7可見,隨著Co(Ⅱ)初始濃度的增加,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的平衡吸附量逐漸增大,25 mg/L時平衡吸附量最大,為92 mg/g。
為進一步研究吸附模式,采用Langmuir吸附等溫模型(式(4))和Freundlich吸附等溫模型(式(5))[15-16]對圖7中的實驗數(shù)據(jù)進行擬合,結(jié)果列于表1。表1表明,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附更符合Langmuir吸附等溫模型(R2=0.991),說明吸附過程以單層吸附為主。采用Langmuir吸附等溫模型可得,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的理論最大吸附量為106 mg/g。

表1 Langmuir和Freunlich等溫吸附模型參數(shù) Table 1 Parameter of Langmuir and Freunlich isotherms model
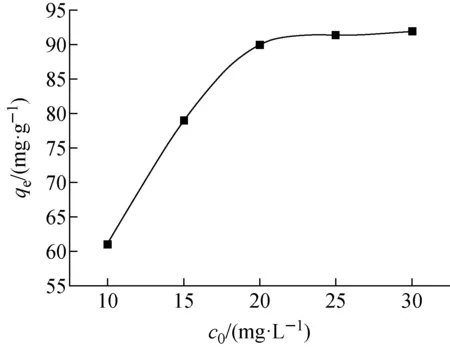
圖7 Co(Ⅱ)初始濃度對Fe3O4@SiO2@UiO-66-SO3H 吸附Co(Ⅱ)的影響Fig.7 Effect of initial concentration of Co(Ⅱ) on adsorption of Co(Ⅱ) on Fe3O4@SiO2@UiO-66-SO3H
(4)
(5)
式中:ce為平衡濃度,mg/L;qm為理論最大吸附量,mg/g;KL為Langmuir吸附平衡常數(shù),L/mg;KF(mg/(g·(L/mg)1/n))和n分別為與吸附能力和吸附強度相關(guān)的Freundlich常數(shù)。
4) 溫度
c0=10 mg/L、t=24 h、初始pH=8.3、m=10 mg下,溫度對Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)的影響示于圖8。由圖8可見,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附量隨溫度的升高而增加,在288、298、308 K下平衡吸附量分別85、92、103 mg/g,表明升溫有利于Co(Ⅱ)的吸附。

圖8 溫度對Fe3O4@SiO2@UiO-66-SO3H 吸附Co(Ⅱ)的影響Fig.8 Effect of temperature on adsorption of Co(Ⅱ) on Fe3O4@SiO2@UiO-66-SO3H
為進一步研究吸附過程的熱力學參數(shù),對圖8中的實驗數(shù)據(jù)進行擬合,采用式(6)、(7)[17]計算其焓變(ΔH?,kJ/mol)、熵變(ΔS?,J/mol/K)和吉布斯自由能變(ΔG?,kJ/mol):
(6)
ΔG?=ΔH?-TΔS?
(7)
式中:T為絕對溫度,K;R為理想氣體常數(shù),取8.314 J/(mol·K);Kd為熱力學常數(shù),其值由Khan[18]公式計算,即以ln(qe/ce)對qe作圖,qe=0時的截距即為lnKd的值。
Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)的熱力學參數(shù)列于表2。由表2可見,ΔG?<0 kJ/mol,說明吸附過程是自發(fā)的;ΔH?>0,說明吸附過程吸熱。綜上可知,F(xiàn)e3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附是一自發(fā)吸熱過程。

表2 Fe3O4@SiO2@UiO-66-SO3H 對Co(Ⅱ)的吸附熱力學參數(shù)Table 2 Thermodynamics parameter for Co(Ⅱ) adsorption on Fe3O4@SiO2@UiO-66-SO3H
2.3 Fe3O4@SiO2@UiO-66-SO3H的循環(huán)利用
c0=10 mg/L、t=24 h、初始pH=8.3、m=10 mg、T=298 K下,F(xiàn)e3O4@SiO2@UiO-66-SO3H的循環(huán)再生性能示于圖9,實驗中采用0.1 mol/L硝酸作洗脫劑。由圖9可知,F(xiàn)e3O4@SiO2@UiO-66-SO3H循環(huán)使用5次后,平衡吸附量從60 mg/g下降至48 mg/g,可能是因為包裹在Fe3O4@SiO2表面的UiO-66-SO3H有微量脫落以及多次循環(huán)吸附后Fe3O4@SiO2@UiO-66-SO3H的量略有減少所致。
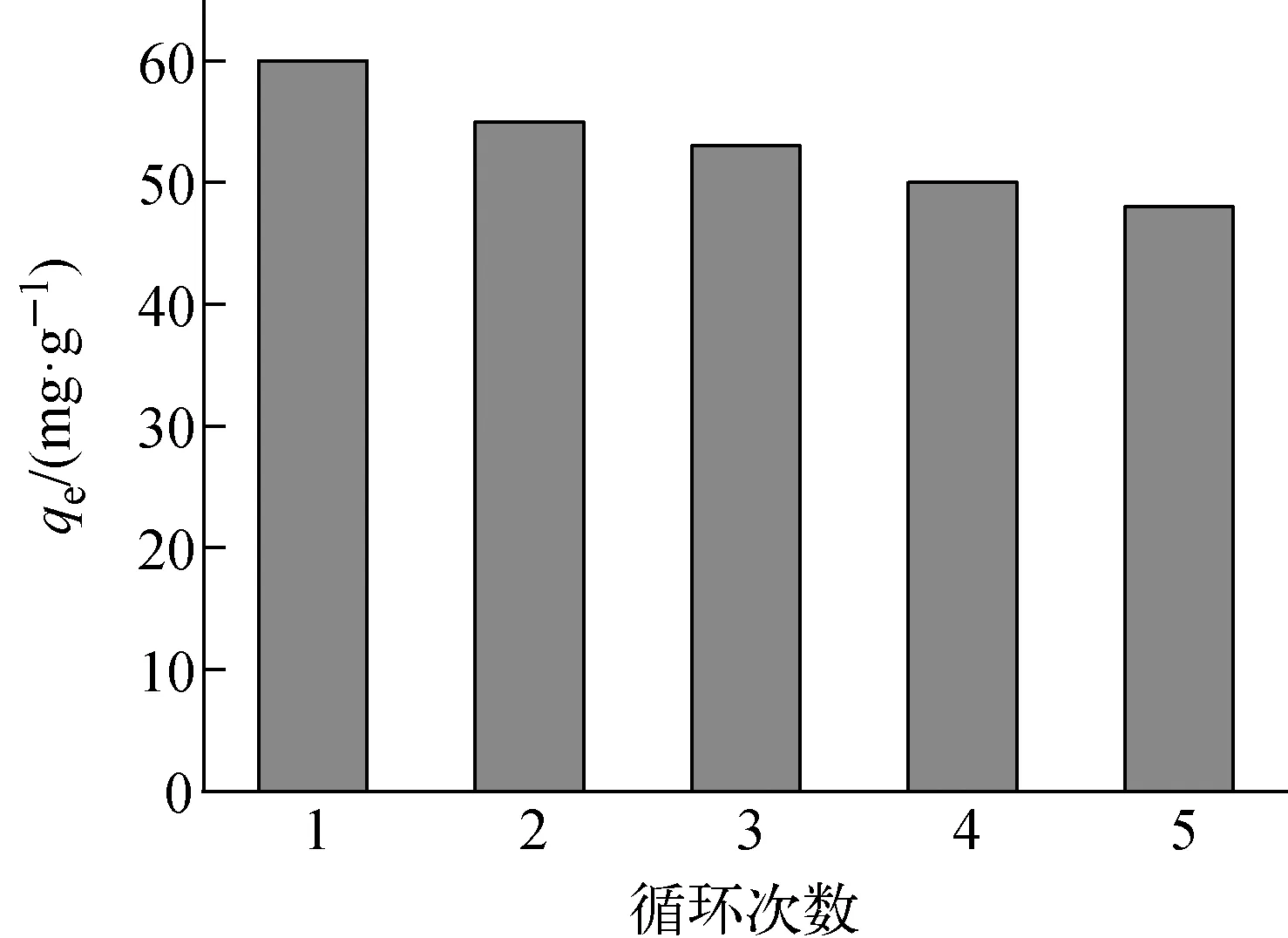
圖9 Fe3O4@SiO2@UiO-66-SO3H的循環(huán)再生性能Fig.9 Recycling performance of Fe3O4@SiO2@UiO-66-SO3H
2.4 吸附機理
Fe3O4@SiO2@UiO-66-SO3H吸附Co(Ⅱ)前后的XPS掃描全譜和元素分譜示于圖10a、b。由圖10a、b可知,Co(Ⅱ)被成功吸附到Fe3O4@SiO2@UiO-66-SO3H上。
Fe3O4@SiO2@UiO-66-SO3H吸附前后S的2p譜示于圖10c。由圖10c可見,吸附后結(jié)合能由168.2 eV變?yōu)?68.4 eV,表明磁性MOFs材料中磺酸基中的S與Co發(fā)生了配位反應(yīng)[19]。169.8 eV處是2-環(huán)己胺基乙磺酸中的S[20],表明一定量的2-環(huán)己胺基乙磺酸被吸附在此吸附劑上。綜上所述,整個吸附過程是一種配位效應(yīng),主要結(jié)合位點是磁性Fe3O4@SiO2@UiO-66-SO3H上的S原子。

a——XPS掃描全譜;b——Co 2p 譜;c——S 2p 譜圖10 Fe3O4@SiO2@UiO-66-SO3H吸附前后的XPS譜Fig.10 XPS spectra of Fe3O4@SiO2@UiO-66-SO3H before and after adsorption
3 結(jié)論
1) 通過水熱法成功將磺酸基功能化的MOFs材料UiO-66-SO3H與SiO2包裹的磁基體Fe3O4相結(jié)合,制得磁性MOFs材料Fe3O4@SiO2@UiO-66-SO3H。
2) FT-IR、XRD、SEM、BET、VSM等的表征結(jié)果表明,所制得的Fe3O4@SiO2@UiO-66-SO3H具有高比表面積、磺酸基官能團、多孔結(jié)構(gòu)、超順磁性、高穩(wěn)定性。
3) Fe3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)吸附效果明顯,在c0=25 mg/L、t=24 h、溫度298 K、初始pH=8.3、m=10 mg下,平衡時吸附量為92 mg/g;Fe3O4@SiO2@UiO-66-SO3H對Co(Ⅱ)的吸附符合準二級動力學模型和Langmiur模型,屬于自發(fā)的吸熱過程;XPS分析證明,吸附劑對Co(Ⅱ)優(yōu)異的吸附能力主要歸因于配位效應(yīng)。因此,F(xiàn)e3O4@SiO2@UiO-66-SO3H作為一種磁性吸附材料,具有良好的吸附性能,易于利用外加磁場實現(xiàn)Co(Ⅱ)吸附后的固-液分離。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網(wǎng)絡(luò)安全與數(shù)據(jù)管理(2022年1期)2022-08-29 03:15:20
導(dǎo)航定位學報(2022年4期)2022-08-15 08:27:00
中學生數(shù)理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數(shù)學備考)(2021年9期)2021-11-24 01:14:36
成都醫(yī)學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數(shù)學備考)(2020年9期)2021-01-04 00:25:14
中學生數(shù)理化·七年級數(shù)學人教版(2020年10期)2020-11-26 08:24:50
數(shù)學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19