離子選擇電極標準加入法測定磷酸中的氯離子
2021-09-28 02:19:06王連軍
磷肥與復肥 2021年8期
關鍵詞:實驗
王連軍
(中國-阿拉伯化肥有限公司,河北 秦皇島 066003)
復合肥料生產中,原料磷酸中的氯離子會阻礙金屬表面鈍化膜的形成,進而導致磷酸輸送、貯存及反應設備快速腐蝕,因此,需嚴格控制磷酸中氯離子含量。磷酸成分較復雜,滴定法測定其中氯離子時干擾離子影響滴定終點的判定,比色法和比濁法在測定色度較深及氯離子含量偏高的磷酸樣品時結果偏差較大。筆者采用氯離子選擇電極標準加入法[1]測定磷酸中氯離子含量,優化測定條件,實現磷酸中氯離子含量的準確、快速測定。
1 實驗方法
1.1 儀器和試劑
儀器:電位測定儀PHS-3C;氯離子選擇電極;飽和甘汞電極;電磁攪拌器。
試劑:氯離子標準溶液(1 mg/mL),稱取經550℃烘干至恒質量的基準氯化鈉1.648 7 g,加水溶解并稀釋至1 L,混勻;硝酸溶液,1+15;氫氧化鈉溶液40 g/L;甲基紅指示液,1 g/L。
1.2 分析步驟
1.2.1 試樣溶液的制備
稱取磷酸試樣0.5~1.0 g(精確至0.000 2 g)于100 mL容量瓶中,加入15 mL硝酸溶液,常溫振蕩溶解10 min,用水稀釋至刻度,混勻。
1.2.2 氯離子的測定
準確吸取含氯離子10~100μg的試樣溶液于50 mL容量瓶中,加入1滴甲基紅指示液,滴加氫氧化鈉溶液至溶液出現混濁并呈黃色,用水稀釋至刻度,混勻,干過濾。準確吸取25 mL濾液于50 mL容量瓶中,逐滴加入硝酸溶液至溶液呈橙紅色(pH為3~4),用水稀釋至刻度,混勻,將其全部轉移至100 mL燒杯中,置于25℃恒溫電磁攪拌器上攪拌。設定電位測定儀測定溫度為25℃,將氯離子選擇電極和飽和甘汞電極插入待測液中,待溶液溫度穩定在25℃,且電極電位讀數穩定后記錄電極電位初讀數E1;然后準確吸取1 mg/kg的氯離子標準溶液0.5 mL加入待測液中,待讀數穩定后記錄電極電位終讀數E2。
氯離子質量分數(w)按式(1)計算:
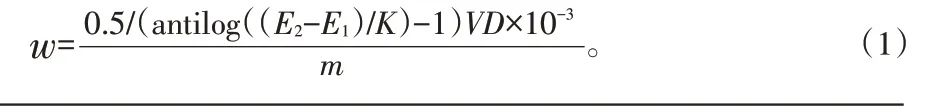
式中 0.5——待測液中加入氯離子標準溶液的質量,mg;
E1——待測液電極電位值,mV;
E2——加入氯離子標準溶液后的電極電位值,mV;
K——能斯特方程斜率的理論值(25℃時為59.16 mV);
D——待測溶液的稀釋倍數;
10-3——質量換算系數;
V——試樣溶液的總體積,mL;
m——試樣的質量,g。
2 實驗結果與討論
2.1 實驗條件的優化選擇
2.1.1 pH適用范圍的選擇
待測試液pH會對離子選擇電極的電位產生影響,而試樣基體的變化及不同的氯離子含量均會影響氯離子選擇電極的pH適用范圍[2],所以待測溶液pH的選擇尤為重要。選取3個不同產地的磷酸樣品,分別調節待測液至不同pH,用離子選擇電極標準加入法測定其氯離子含量,結果見圖1。
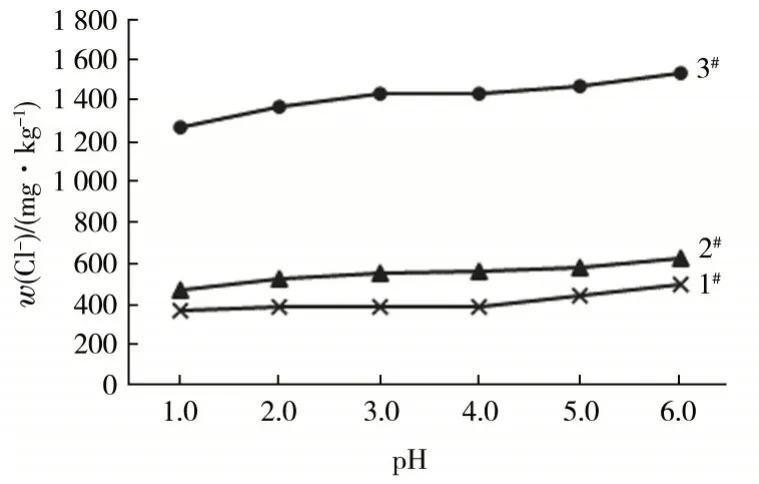
圖1 pH對氯離子測定結果的影響
由圖1可知,當待測試液pH在3~4時,測得氯離子含量趨于穩定。說明在該pH區間,磷酸的基體干擾對氯離子的測定影響較小,有利于保持電極電位穩定,適合磷酸中氯離子的準確測定。
2.1.2 干擾消除與離子強度調節劑的選擇
磷酸中氯離子測定的主要干擾物為鐵、錳等金屬離子,鐵、錳離子可通過沉淀分離或加入檸檬酸鈉等掩蔽消除[3]。考慮到磷酸體系的復雜性,待測液與掩蔽劑、離子強度調節劑等的相互影響,選擇3種方案分別對上述3個磷酸樣品和試劑磷酸進行實驗。方案A,調待測液pH≥4,用水定容至50 mL,過濾,吸濾液25 mL,調pH至3~4,用水定容至50 mL后測定;方案B,調待測液pH至3~4,加硝酸鉀-檸檬酸鈉溶液1 mL,用水定容后測定;方案C,調待測液pH≥4,用水定容,過濾,吸濾液調pH至3~4,加硝酸鉀溶液1 mL,用水定容后測定。實驗結果與比色法結果對比見表1。
由表1可知,方案A的測定結果與比色法最為接近,方案B、C結果與比色法均存在較大正偏差。考慮到試劑純度等因素,實驗中使用了優級純硝酸鉀w(Cl-)≤0.000 1%),并對標明w(Cl-)≤0.003%的試劑磷酸進行對照測定,結果上述問題仍然存在。說明方案A即通過沉淀消除鐵、錳離子干擾后,在pH為3~4的水溶液體系中采用離子選擇電極標準加入法可準確測定磷酸中氯離子含量。
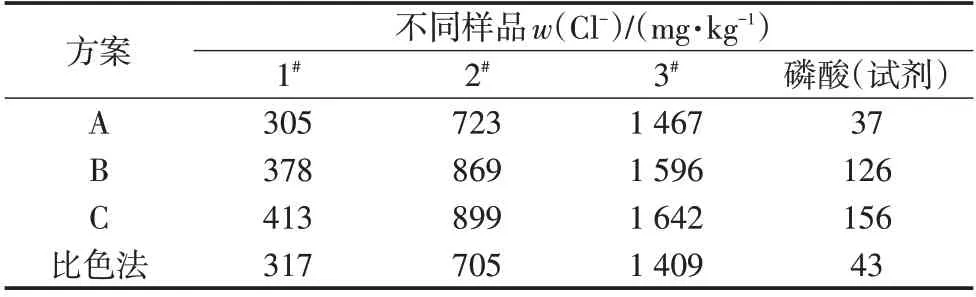
表1 各方案實驗結果與比色法對比
2.2 精密度實驗
選取上述3個磷酸樣品,用離子選擇電極標準加入法分別測定氯離子含量,每個樣品重復測定6次,結果見表2。
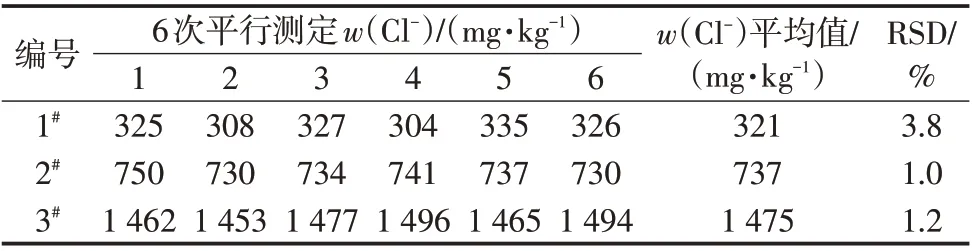
表2 精密度實驗結果
由表2可知,該方法相對標準偏差在1.0%~3.8%,結果重現性較好,滿足精密度要求。
2.3 加標回收實驗
稱取不同氯離子含量的3個磷酸樣品,加入氯離子標準溶液,按上述實驗方法分別測定氯離子含量并計算加標回收率,結果見表3。
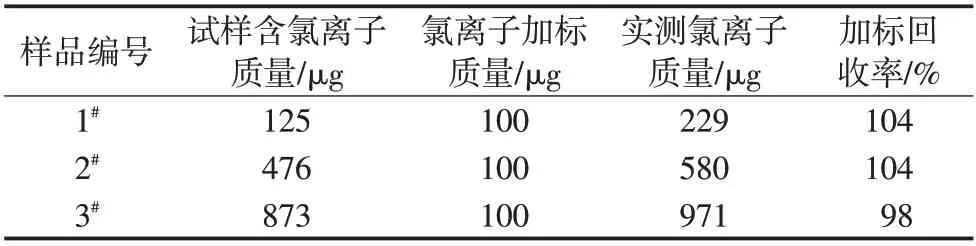
表3 加標回收實驗結果
由表3可知,3個磷酸樣品加標回收率在98%~104%,說明該方法測定磷酸中氯離子含量滿足定量分析的準確度要求。
3 結論
先調節待測液pH≥4沉淀分離磷酸中鐵、錳等干擾離子,再調濾液pH至3~4,用氯離子選擇電極標準加入法測定磷酸中的氯離子含量。該方法可有效消除磷酸的基體干擾,操作簡便,結果準確可靠。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
