高效液相色譜法測定小麥粉中玉米赤霉烯酮的不確定度評定
2021-09-01 12:34:14◎趙飛
現代食品 2021年14期
◎ 趙 飛
(遼寧省檢驗檢測認證中心,遼寧 沈陽 110000)
測量不確定度評定是按照實際檢測過程和相關信息對檢測結果的分散性進行評估的一種方式[1]。測量不確定度可以客觀的評價檢測數據,尤其是當檢測數據處于臨界值時,進行不確定度評定可以保障檢測結果的準確性[2]。
玉米赤霉烯酮,又稱F-2毒素,是玉米赤霉菌的代謝產物[3-4]。玉米赤霉烯酮易污染玉米、小麥等谷物,尤其是當這些谷物儲存不當時更易被污染[5]。它對神經系統、心臟、腎臟等有毒害作用。因此本文嚴格按照《食品安全國家標準 食品中玉米赤霉烯酮的測定》(GB 5009.209—2016)[6]操作,對其檢測結果的不確定度進行評估,為實驗室出具的玉米赤霉烯酮檢測結果的準確性提供科學依據。
1 材料和方法
1.1 儀器與試劑
Agilent 1260 Infinity液相色譜儀(美國安捷倫公司)、ZORBAX SB-C18色譜柱(4.6 mm×150 mm,0.5 μm,美國安捷倫公司)、BSA224S-CW電子天平(德國賽多利斯公司)、GM200高速粉碎機(德國萊馳公司)、FJ300-SH均質器(德國IKA公司)、TTL-DCII氮吹儀(北京同泰聯科技發展有限公司)、X1R高速冷凍離心機(德國Thermo公司)。
乙腈、甲醇(色譜純,美國Fisher公司);氯化鈉、氯化鉀、磷酸氫二鈉、磷酸二氫鉀(分析純,國藥集團化學試劑有限公司);水,均為一級水;玉米赤霉烯酮標準物質(濃度為100.2 μg·mL-1,ROMER公司)。
1.2 實驗方法
1.2.1 標準溶液的配制
準確吸取1.0 mL玉米赤霉烯酮標準溶液于10 mL容量瓶中,用乙腈稀釋配制成濃度為1.02 μg·mL-1的標準儲備液,-18 ℃避光保存。用70%乙腈溶液將上述標準儲備液稀釋成系列標準溶液,濃度分別為10 ng·mL-1、25 ng·mL-1、50 ng·mL-1、75 ng·mL-1和100 ng·mL-1,標準系列溶液臨用現配。
1.2.2 液相色譜參考條件
色 譜 柱:ZORBAX SB-C18色 譜 柱(4.6 mm×150 mm,0.5 μm);流動相:乙腈∶水=70∶30(v/v);流速:0.7 mL·min-1;柱溫:30 ℃;激發波長:274 nm;發射波長:440 nm;進樣體積:100 μL。
1.2.3 樣品前處理
稱取40 g試樣(精確至0.1 g)于均質杯中,加入4 g氯化鈉、100 mL提取液,高速攪拌提取2 min,過濾。移取10 mL濾液于40 mL水中,經玻璃纖維濾紙過濾至澄清;準確移取10 mL上述濾液至免疫親和柱上,直至有部分空氣進入,用5 mL水淋洗柱子一次,再準確加入1.5 mL甲醇洗脫,流速為1滴/s。收集洗脫液于玻璃試管中,55 ℃下氮氣吹干,用1.0 mL流動相溶解殘渣,過膜待測。
1.2.4 數學模型的建立
樣品中玉米赤霉烯酮的含量按式(1)計算。
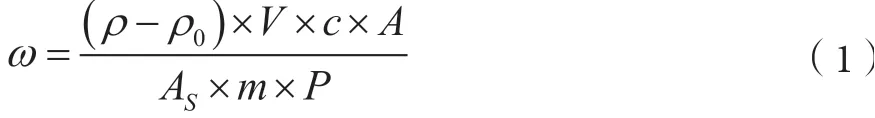
式中,ω-樣品中玉米赤霉烯酮的含量,μg·kg-1;ρ-由標準曲線得出的樣品溶液中玉米赤霉烯酮的濃度,ng·mL-1;ρ0-空白溶液中玉米赤霉烯酮的濃度,ng·mL-1;V-樣品定容體積,mL;c-系數;A-試樣溶液中玉米赤霉烯酮的色譜峰面積;AS-標準溶液中玉米赤霉烯酮的色譜峰面積;m-稱樣量,g;P-加標樣品回收率。
2 標準不確定度分量的評定
2.1 標準品引入的不確定度ur(s)
2.1.1 標準品純度引入的不確定度ur(c1)
通過查看玉米赤霉烯酮的標準物質證書,得到該標準品相對不確定度U=0.2%,k=2,則其相對標準不確定度為:
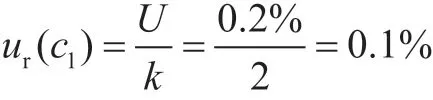
2.1.2 標準儲備液配制引入的不確定度ur(c2)
標準儲備液配制中用到1 mL的A級單標線吸量管、10 mL容量瓶,允許誤差分別為±0.008 mL、±0.020 mL,按照均勻分布考慮,取則吸量管和容量瓶引入的不確定度為:
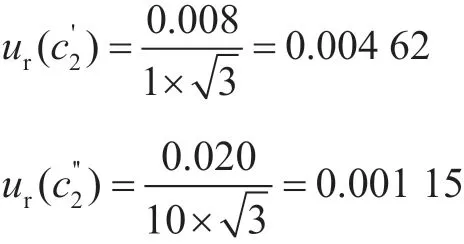
標準溶液配制室的溫度通常控制在(20±5)℃,20 ℃下乙腈的膨脹系數為1.37×10-3℃-1,由實驗室溫度變化引起體積變化的相對標準不確定度為:
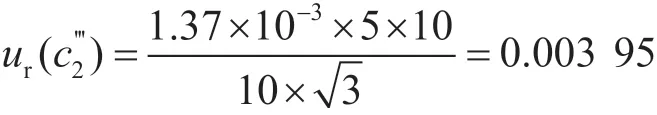
綜上,標準儲備液配制過程引入的不確定度為:
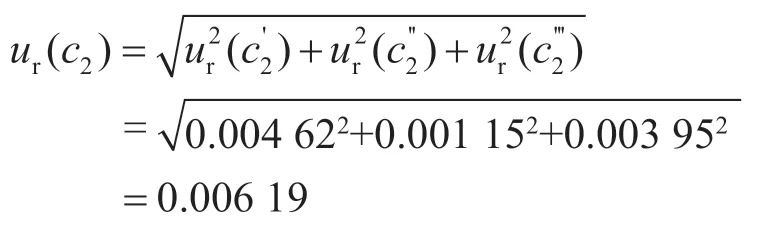
2.1.3 標準工作液配制引入的不確定度ur(c3)
標準曲線配制過程中各分量的相對合成標準不確定度見表1。
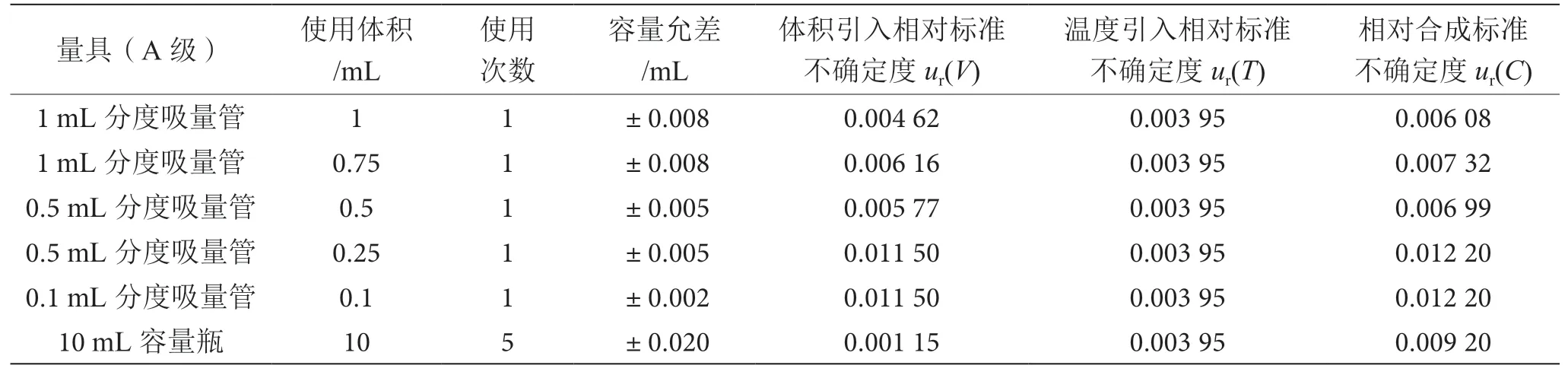
表1 標準曲線配制過程引入的不確定度表
根據各分量,標準曲線配制過程引入的相對標準不確定度為:
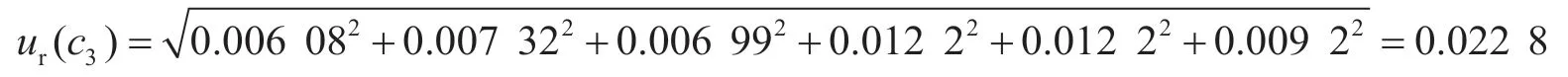
2.1.4 標準曲線擬合引入的不確定度ur(c4)
本方法對濃度依次為10 ng·mL-1、25 ng·mL-1、50 ng·mL-1、75 ng·mL-1和100 ng·mL-1的玉米赤霉烯酮標準溶液進行測定,以玉米赤霉烯酮標準溶液的濃度為橫坐標,以響應值峰面積為縱坐標進行線性擬合,校準曲線方程見表2,并對陰性樣品加標進行6次平行測定,測得結果分別為52.0 ng·mL-1、52.6 ng·mL-1、53.2 ng·mL-1、53.3 ng·mL-1、53.0 ng·mL-1和52.8 ng·mL-1,平均值為52.8 ng·mL-1,由標準曲線引入的不確定度可由式(2)計算。
表2 碘化鉀飽和溶液的放置時間對測定結果的影響表

表2 玉米赤霉烯酮校準曲線表
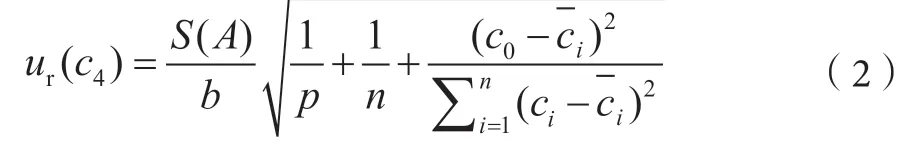
式中,ur(c4)-曲線擬合引入的不確定度;S(A)-標準溶液待測物質信號殘差的標準差;b-校準曲線的斜率;p-樣品溶液檢測次數(6次);n-標準溶液的測定次數(5次);c0-校準曲線校正后待測樣品中玉米赤霉烯酮濃度的平均值,ng·mL-1;ci-標準曲線各點玉米赤霉烯酮濃度理論值,ng·mL-1;標準曲線各點玉米赤霉烯酮濃度的平均值,ng·mL-1。
其中,S(A)可按公式(3)計算。
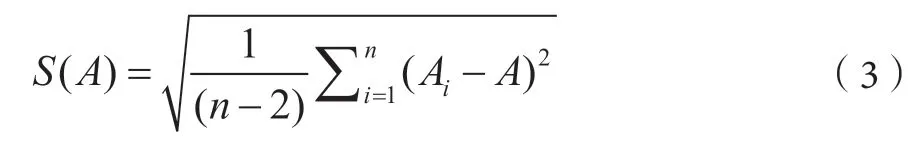
式中,n-校準曲線的點數;Ai-標準溶液各濃度點的峰面積;A-根據標準曲線算出的理論峰面積。
由校準曲線擬合引入的相對標準不確定度為ur(c4)=0.001 88。
綜上,標準物質引入的相對標準不確定度計算為:
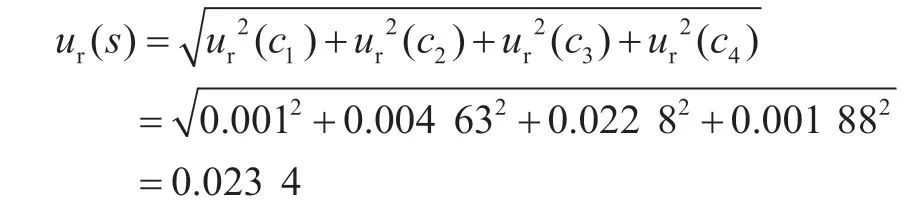
2.2 樣品稱量引入的不確定度ur(m)
采用分度為0.1 g的天平稱量待測樣品,該天平在0~50 g稱量范圍內的最大允許誤差為±0.05 g,樣品稱量過程引入的相對不確定度為:
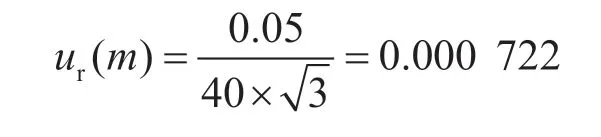
2.3 樣品前處理引入的不確定度ur(V)
樣品前處理過程中采用100 mL量筒,其容量允差為±1.0 mL,在(20±5)℃條件下,由此引入的相對標準不確定度為:
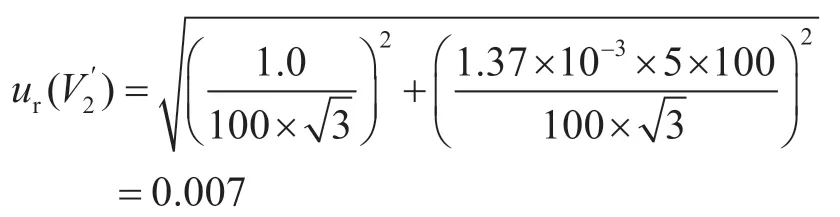
移取10 mL提取液的濾液,并加入40 mL水稀釋混勻,10 mL單標線吸量管和50 mL刻度吸量管的容量允差為±0.020 mL、±0.10 mL,稀釋過程引入的相對標準不確定度為:
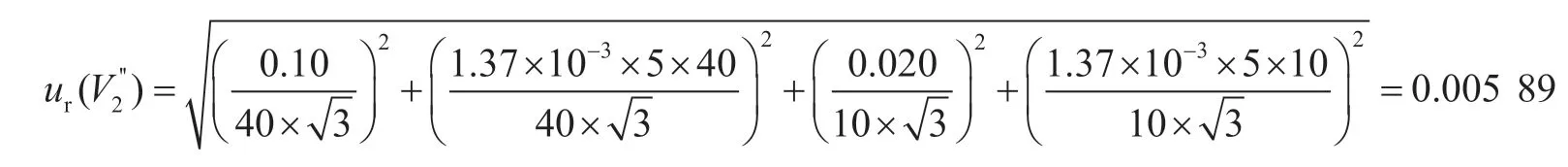
準確移取10 mL濾液至免疫親和柱上凈化,最后用1.0 mL流動相復溶,10 mL和1 mL單標線吸量管的容量允差為±0.020 mL、±0.007 mL,稀釋過程引入的相對標準不確定度為:
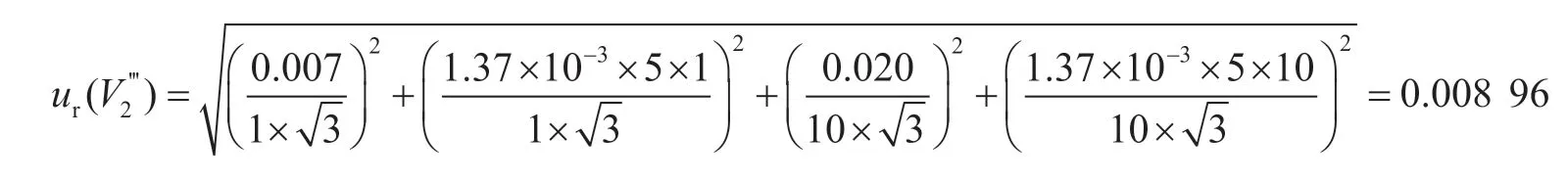
該過程引入的相對合成標準不確定度為:
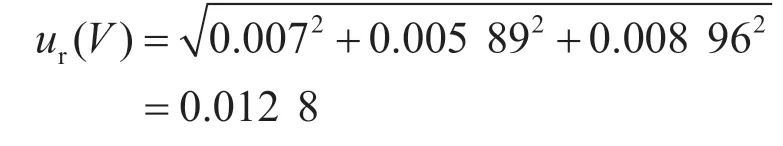
2.4 重復性引入的不確定度ur(R)
開展加標實驗,添加量為70.0 μg·kg-1,測定值分別為65.0 μg·kg-1、65.7 μg·kg-1、66.5 μg·kg-1、66.6 μg·kg-1、66.2 μg·kg-1和66.0 μg·kg-1,平均值為66.0 μg·kg-1。按照A類評定,應用貝塞爾公式計算,待測物的實際標準偏差為0.590,其相對標準不確定度為:
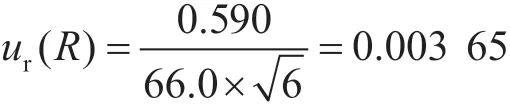
2.5 回收率引入的不確定度ur(P)
向不含玉米赤霉烯酮的小麥粉中加標,添加量為70.0 μg·kg-1,通過前處理并檢測計算,得回收率分別為92.9%、93.9%、95.0%、95.1%、94.6%和94.3%,平均值為94.3%,按照A類評定應用貝塞爾公式計算待測物的實際標準偏差為0.843%,其標準不確定度為:
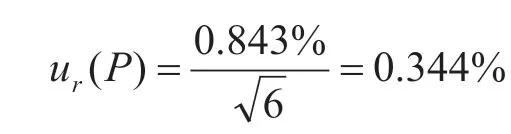
用t來檢驗確定平均回收率是否與1有顯著差異,當n=6,自由度為5時,在置信概率為95%時,t0.95(5)=2.571,該回收率下即t>2.571,需要用回收率對結果進行修正。回收率引入的相對標準不確定度為:
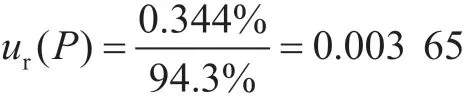
3 合成不確定度
采用本方法檢測小麥粉中玉米赤霉烯酮的相對標準不確定度為:
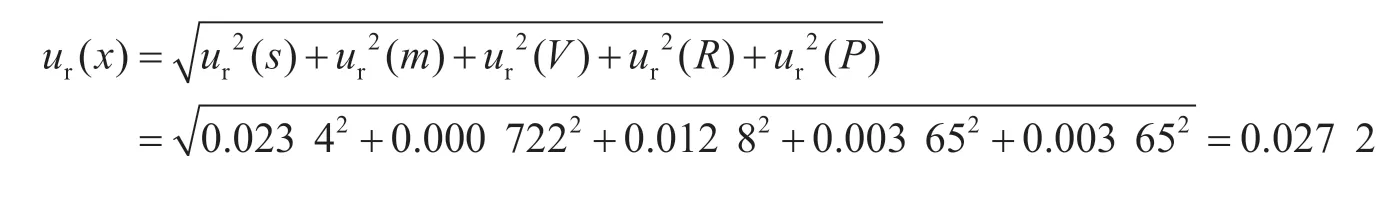
玉米赤霉烯酮的合成標準不確定度為:
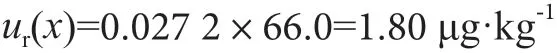
4 擴展不確定度的計算和檢測結果的表示
在95%置信概率下,擴展因子k=2,則采用本方法檢測小麥粉中玉米赤霉烯酮的擴展不確定度為U95=k×ur(x)=3.60 μg·kg-1,小麥粉中玉米赤霉烯酮的檢測結果表示為(66.0±3.60)μg·kg-1;k=2。
5 結論
采用食品安全國家標準方法測定小麥粉中玉米赤霉烯酮含量,對檢測結果的不確定度進行評定。通過評定,該實驗中標準曲線配制過程引入的不確定度貢獻最大。因此,在實驗室采用該方法開展玉米赤霉烯酮檢測時,要注意選用合適的量具,并定期開展檢定校準,嚴格控制標準溶液配制室溫度,切實減小不確定度分量,保證檢測數據的科學性和合理性。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
