山東地區1056例新生兒聽力篩查聯合耳聾基因篩查結果分析
2021-09-01 01:25:46孫毅劉雅琳劉曉莉孫麗麗潘持國張斌
中華耳科學雜志 2021年4期
孫毅 劉雅琳 劉曉莉 孫麗麗 潘持國 張斌
山東省康復研究中心(山東省康復醫院)(濟南250109)
耳聾是由遺傳因素或環境因素導致的生理機能缺陷,嚴重影響人類健康和生活質量。耳聾可以由單個基因位點突變或多基因位點突變引起,也可以由環境因素或基因和環境因素共同導致[1]。研究發現65%的耳聾是由遺傳因素引起的[2]。隨著對新生兒聽力篩查工作的開展,常規聽力學篩查逐漸顯現一定的局限性,不能及時發現藥物性耳聾和遲發性耳聾。因此,遺傳性耳聾基因篩查聯合聽力篩查尤為重要。本研究利用NGS Panel技術對山東地區新生兒進行遺傳性耳聾基因檢測,相比于質譜檢測方法和微陣列芯片檢測方法,該方法可覆蓋更多致聾基因和致聾位點,有利于實現早預防、早發現、早干預、早康復的長效機制,為政府制定聽力殘疾防控體系提供科學依據,最終達到防聾減殘、提高人口素質的目標[3]。
1 對象和方法
1.1 研究對象
研究對象樣本主要來源于2019年4月至2019年10月,在山東省內各地分娩的新生兒1056例,篩查前已明確告知聽力篩查和遺傳性耳聾基因檢測的相關知識,監護人或家屬簽訂知情同意書。
1.2 聽力檢測方法
新生兒出生2-3天后,在自然睡眠狀態或喂奶安靜狀態下,在環境噪聲小于40dB(A)的房間內,分娩醫院使用耳聲發射(Oto-acoustic Emissions,OAE)進行聽力篩查,由儀器自動判讀“通過”或“不通過”,聽力初篩未通過的新生兒,需要42天后進行復篩。復篩未通過會將檢測結果告知家長,并要求3月齡時進行聽力學診斷。
本研究聽力損失程度以聽性腦干反應(audi‐tory brainstem response,ABR)檢測結果為參照標準進行判定。ABR波Ⅴ反應閾值≤30dB nHL為聽力正常的標準,波Ⅴ反應閾值為31~ 50dB nHL視為輕度聽力損失,波Ⅴ反應閾值為51~ 70dB nHL視為中度聽力損失,波Ⅴ反應閾值為71~ 90dB nHL視為重度聽力損失,波Ⅴ反應閾值>90dB nHL視為極重度聽力損失。
1.3 耳聾基因檢測方法
新生兒出生2天后,由各助產機構使用特定采血卡,采集新生兒足跟血2個血斑,每個血斑直徑不得小于8mm,常溫下自然晾干,分別裝入自封袋內。實驗室負責對標本質量進行驗收,驗收合格后郵寄至北京博奧醫學檢驗所有限公司進行18個遺傳性耳聾基因100個位點NGS Panel檢測,并在10個工作日內將檢測結果反饋至新生兒出生分娩機構。最后聯合聽力篩查結果和遺傳性耳聾基因檢測結果,隨訪新生兒聽力損失發生情況。
1.4 檢測流程
北京博奧醫學檢驗所有限公司實驗室首先進行核酸提取,使用多重PCR技術一次性對GJB2,SLC26A4,GJB3,MYO15A,TECTA,DIABLO,COCH,DSPP,GPR98,DFNA5,TMC1,MT-CO1,MT-RNR1,MT-TH,MT-TS1,MT-TL1,PRPS1,MYO7A,18個致聾基因的100個已知致病位點進行檢測。對于陽性結果,使用Sanger測序復核結果后再進行報告發放。
2 結果
2.1 聽力檢測結果分析
在1056例新生兒中,其中聽力篩查未通過者5人(0.47%),經聽力學診斷確診聽力損失患兒3人(見表2),占篩查未通過的60.00%。其中,1例右耳中度聽力損失,左耳輕度聽力損失,另外2例單獨左耳輕度聽力損失。
2.2 基因檢測結果分析
對1056例新生兒全部進行18個遺傳性耳聾基因100個位點檢測,發現耳聾基因突變者77例,檢出率為7.29%(見表1)。通過檢測結果分析顯示SLC26A4基因雜合突變檢出37例,檢出率為3.50%;GJB2基因突變檢出30例,檢出率2.84%,其中雜合突變29例,復合雜合突變1例;GJB3基因雜合突變檢出8例,檢出率0.76%;線粒體12SrRNA基因均質突變1例,檢出率0.10%。SLC26A4基因突變合并線粒體12SrRNA基因突變的雙基因突變檢出1例。在3個聽力損失患兒中遺傳性耳聾基因檢測陽性2例,分別為SLC26A4基因c.2168A>G雜合突變和GJB2基因c.176-191del16/c.253T>C復合雜合突變,陰性1人。
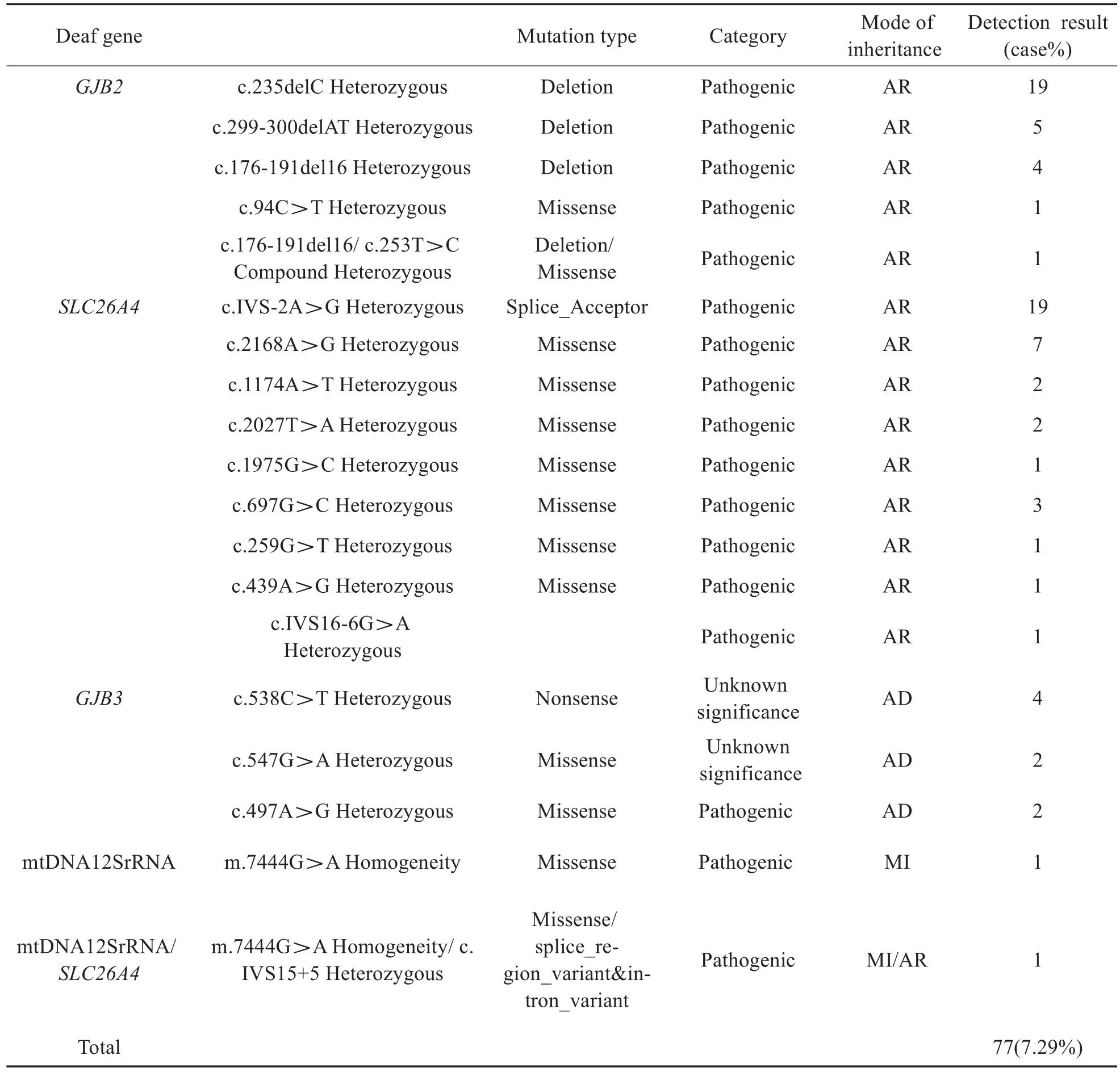
表1 1056例新生兒中100個致聾基因突變的攜帶率Table 1 Carrier frequency of 100 hotspot mutations 1056 randomly-selected cases
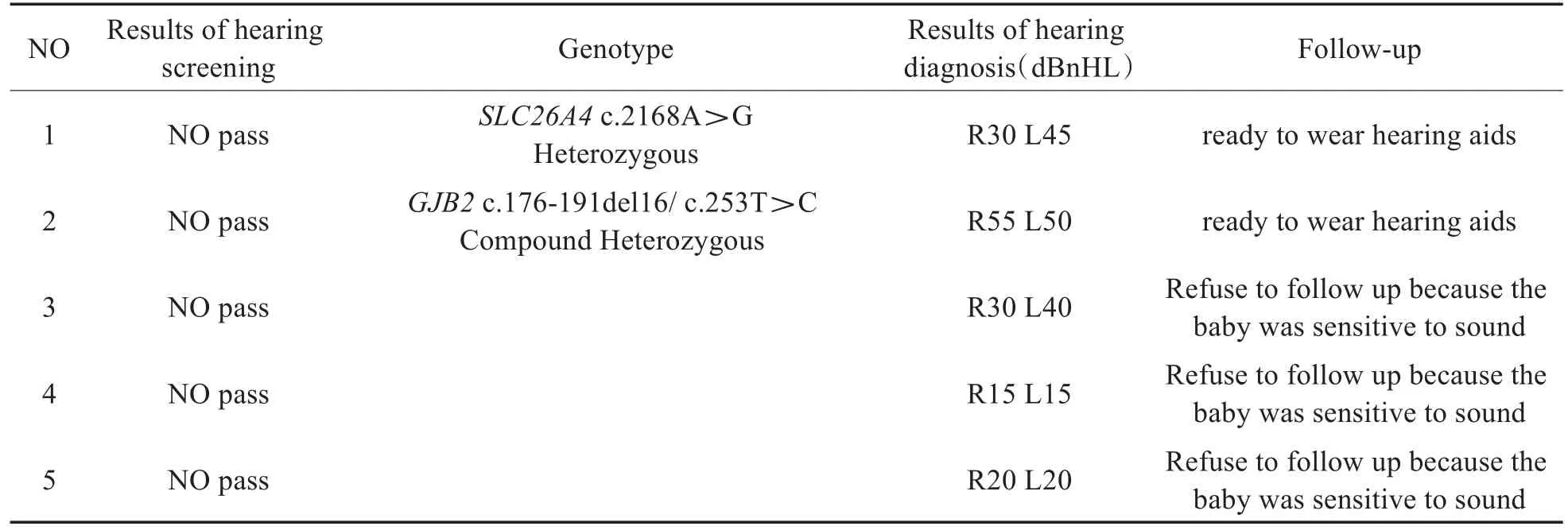
表2 兒童基本信息及聽力結果Table 2 Demographic information and audiological diagnoses of subjects
3 討論
耳聾是影響人類健康和造成人類殘疾的常見疾病。目前研究主要針對少數常見的與耳聾相關的基因中有限的熱點突變位點,只有少數能通過檢測尋找突變[4]。本研究采用NGS Panel的方法,檢測與耳聾相關的18個遺傳性耳聾基因中的100個致聾位點。如此大范圍的基因檢測覆蓋率有效的彌補了常規檢測方法的不足,不僅對耳聾相關基因突變頻率的預期更加準確,也可以為醫務工作者對患者明確病因提供更加可靠的檢測結果。
3.1 耳聾基因攜帶者檢出結果分析
本研究共檢測山東地區新生兒1056例,耳聾基因篩查突變者77例,檢出率7.29%。高敏等對1737名孕婦進行18個遺傳性耳聾基因100個致聾位點檢測,發現遺傳性耳聾攜帶率7.25%[5],本研究結果與其報道基本一致。Shui lian Chen等對近年來全國各地區報道的18篇新生兒遺傳性耳聾基因檢測的文獻進行meta分析,發現全國各地新生兒遺傳性耳聾基因攜帶率為4.70%[6]。王秋菊等對近年來全國出生的110余萬例新生兒進行遺傳性耳聾基因檢測結果分析,發現遺傳性耳聾基因在新生兒中的攜帶率為4.78%[7]。兩項研究結果表明,在我國常見的遺傳性耳聾基因攜帶率基本一致。在此之前,山東地區學者報道了本地的遺傳性耳聾基因攜帶率,孫磊報道山東省濟寧市新生兒耳聾基因陽性檢出率為5.60%[8]。本研究的遺傳性耳聾基因陽性檢出率高于孫磊報道的5.60%,原因可能是檢測的目的致聾基因數量、方法及突變位點存在差異。查閱文獻發現,目前我國最常見的耳聾基因檢測為9項、15項耳聾基因芯片檢測和20項飛行時間質譜檢測。每個地區檢測結果顯示,GJB2基因與SLC26A4基因變異攜帶率較接近[9]。參考孫磊報道數據(SLC26A4基因突變攜帶率2.19%),發現本研究的SLC26A4基因檢出率增加最明顯,增加率占總檢出率的17.97%(1.31%/7.29%)。孫磊、王秋菊等的檢測方法為飛行時間質譜技術和耳聾基因芯片技術,對中國人常見GJB2、GJB3、SLC26A4和線粒體12srRNA等4個耳聾易感基因進行檢測。本研究的檢測方法為NGS Panel法,對18個遺傳性耳聾基因100個致聾位點進行檢測。因此造成本研究遺傳性耳聾基因攜帶檢出率高于孫磊等報道的5.60%及王秋菊等報道的4.78%。
3.2 遺傳性耳聾基因攜帶者等位基因檢出率分析
依據檢測結果中遺傳性耳聾基因攜帶者等位基因分析,本研究中SLC26A4基因的c.IVS7-2A>G位點突變檢出率最高,為1.80%(19/1056),其次分別為c.2168A>G 位點0.66%(7/1056),c.697G>C位點 0.28%(3/1056),c.1174A>T、c.2027T>A 位點0.19%(2/1056),c.1975G>C、c.439A>G、c.259G>T、IVS16-6G>A位點0.10%(1/1056)。學者研究報道,SLC26A4基因含有21個外顯子,在我國,約96%的前庭水管擴大由此基因突變致病,在歐美國家僅有40%左右。說明在中國人群中SLC26A4基因型與表型關系更密切。許暉雁等的研究分析表明此基因的突變患者人工耳蝸術后聽力言語康復效果普遍較好,并且SLC26A4基因突變是獨立的預后因素[10]。在中國聾病人群中最常見的突變位點為 c.IVS7-2A>G,其次為 c.2168A>G[11]。2019 年Mahbobeh等對中東伊朗地區聾人基因檢測發現,SLC26A4基因的檢出率為6.39%,其中以c.1334T>G位點最為常見[12]。因此表明SLC26A4基因位點突變存在著明顯的地域和種族差異。本研究結果與我國前人研究結果一致,主要突變位點為c.IVS7-2A>G。
GJB2基因突變發生頻率最高位點是c.235delC,突變率為1.80%(19/1056),其它位點依次 為 c.299_300delAT位 點 0.47%(5/1056)、c.176del16位點0.38%(4/1056)、c.94C>T位點0.10%(1/1056)。GJB2基因突變在北歐猶太教徒、高加索人中,出現頻率較多的基因位點突變分別是c.35delG、c.167delT 和 c.235delC[13]。 在 亞 洲 ,c.235delC是最常見的位點突變,突變率為5.22%,戴樸等在2009年對2063例中國非綜合征性耳聾患者進行GJB2基因突變頻譜研究發現,c.235delC突變位點最為常見,占GJB2基因致聾突變的68.9%,其次為c.299_300delAT[14]。本研究GJB2基因致聾位點與戴樸等的研究一致,突變頻率最高位點分別為c.235delC和c.299_300delAT。同時本研究發現罕見致病位點c.94C>T[15],但本研究的突變檢出率遠遠低于戴樸等的報道,原因在于本研究的對象為新生兒,前人研究多集中在聽障人群。
3.3 聽力篩查情況及基因陽性聽力學特點
本研究新生兒聽力篩查特點,在1056例新生兒中聽力篩查未通過者5人,占全部人數的0.47%。經聽力學診斷,確診聽力損失患兒3人,其中2人耳聾基因檢測陽性。發現GJB2基因復合雜合突變患者1例,聽力學診斷結果顯示,右耳中度聽力損失,左耳輕度聽力損失,經過隨訪發現聽力保持穩定,未出現聽力再次下降。文獻報道,GJB2基因純合或復合雜合突變的兒童聽力損失程度以重度為主[16],需長期跟蹤隨訪。本研究中3例聽力篩查未通過的新生兒中,有1例通過18個致聾基因檢測,結果未見異常,表明可能存在檢測位點之外的基因突變,所以耳聾基因檢測并不能取代聽力篩查。但若不進行基因檢測,由遺傳性因素導致的耳聾患者不能明確病因,以及耳聾基因攜帶個體不會被跟蹤隨訪,可能會對其父母再生育和他們的后代造成影響。目前耳聾疾病沒有理想的治療措施,但新生兒耳聾基因檢測是聽力篩查的重要補充,聽力篩查與耳聾基因聯合篩查可最大限度地早期發現聾病人群[17]。聽力檢測通過組的2名嬰兒被檢測出帶有MT-CO1:m.7444G>A突變,應一生停用氨基糖苷類抗生素,以避免藥物誘發性聽力受損。特別是我們發現聽力檢測通過組的一名嬰兒同時也帶有其他基因突變。我們建議其盡早注意聽覺易感性變化,并且關注聽力健康。
4 結論
基因突變具有隨機性、不定向性、多害少利等特點,因此,本研究利用NGS Panel檢測技術對山東地區1056例新生兒進行檢測,旨在提高檢測率,了解該地區可能致聾基因攜帶情況,為山東省新生兒耳聾的積極干預和治療提供了指導,為耳聾相關基因突變頻率的準確估計提供了依據。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中國生殖健康(2020年2期)2021-01-18 02:51:26
家庭醫學(下半月)(2019年9期)2019-10-12 08:04:06
家庭醫學(下半月)(2019年8期)2019-09-25 09:02:00
小學生導刊(2018年13期)2018-06-29 03:49:00
媽媽寶寶(2017年3期)2017-02-21 01:22:12
海峽科技與產業(2016年3期)2016-05-17 04:32:12
