多糖的增稠、膠凝及乳化特性研究進展
2021-08-31 03:30:28李秀秀郭玉蓉
食品科學 2021年15期
李秀秀,尚 靜,楊 曦,薛 佳,2,3,4,*,郭玉蓉,3,4
(1.陜西師范大學食品工程與營養科學學院,陜西 西安 710119;2.農業農村部都市農業重點實驗室,上海 200240;3.國家蘋果加工技術研發專業中心,陜西 西安 710119;4.西部果品資源高值利用教育部工程研究中心,陜西 西安 710119)
多糖廣泛存在于自然界中,是最為豐富的天然高分子資源之一[1]。多糖通常是指由多個單糖分子經失水、 縮合而形成的結構復雜且分子質量龐大的一類物質。根據來源及結構特點,可將多糖劃分為植物多糖、 動物多糖、微生物多糖、線性結構多糖、分支化多糖、中性電荷多糖、陰離子多糖、陽離子多糖、親水性多糖、表面活性多糖等[2]。雖然多糖種類繁多,結構差異較大,但從廣義層面而言,凡是符合高分子化合物概念的碳水化合物及其衍生物均可稱為多糖。其中,食品多糖特指食品加工過程中被允許作為添加性成分,用以提高食品感官、質構特性及營養價值的多糖,是食品膠的重要組成部分。因此,食品多糖的范圍相對較窄,某種程度上特指已經商業化的多糖,如果膠[3-4]、結冷膠[5]、瓊脂糖[6]、黃原膠、卡拉膠[7]、淀粉及改性淀粉[8]、纖維素及其衍生物等[2]。除淀粉外,大多數食品多糖不能被人體的消化酶水解,但這些多糖普遍具有膳食纖維的功能價值,可起到降血糖、降血壓、降低膽固醇以及改善人體腸道菌群平衡的作用[9-10]。
多糖本質上屬于天然高分子,因此具有高分子化合物的一般屬性。例如,當多糖完全水化后,其水溶液常表現出各異的流變學特性,包括增稠、膠凝及乳化特性等。一般地,多糖溶液的流變學特性受多糖自身分子結構和外界環境因素的影響。其中,影響多糖流變學特性的結構參數主要包括分子質量、電荷密度、分支化程度等因素,外界環境因素則主要包括pH值、溫度、離子類型和強度、有無其他共溶質存在等。雖然影響多糖流變學特性的因素眾多,且影響途徑不同,但這些因素影響多糖溶液流變學特性的本質在于影響多糖在水溶液中的超分子結構[11-12]。對于典型的凝膠多糖,如結冷膠、卡拉膠、瓊脂糖等,當溶液體系由溶膠態向凝膠態轉變時,多糖分子彼此間往往形成穩定的交聯區,構成空間三維網絡結構[13-14]。當體系由凝膠態向溶膠態轉變時,穩定的分子間交聯重新打開,體系再次以溶膠形式存在,因此這類多糖常常表現出可逆的凝膠化過程。對于非凝膠多糖,如黃原膠、魔芋膠、刺槐豆膠等[15-17],水化后多糖分子傾向于形成非穩定的分子間聚集,這樣的結構容易被外界應力擾亂而解聚集,因此這類多糖形成的溶液具有良好的增稠效果,但膠凝能力較差。此外,一些多糖分子結構中含有少量的疏水官能團,使得它們同時表現出親水和疏水的特性,稱為表面活性多糖或兩親性多糖。表面活性多糖能夠吸附至油-水界面,并具有在油滴表面形成一層水化膜、防止油滴聚集的能力,因此表面活性多糖可作為乳化劑制備乳液[8,15,18-20]。
食品多糖的膠凝、增稠、乳化等流變特性和分子結構密切相關,本質上是多糖超分子結構的宏觀體現[3,20-26]。 因此,理解食品多糖流變學特性的本質需要從解析多糖超分子結構的角度入手。雖然目前國內外有關多糖資源開發利用的文獻已有不少報道[27-30],但鮮有從多糖高分子化合物的本質屬性出發形成專門解讀多糖流變學特性的文獻綜述。另一方面,隨著國內食品工業的快速發展,越來越多的食品多糖被用于改善和優化食品結構,因此對食品多糖流變學本質進行解讀顯得尤為必要。因此,本文探討了食品多糖膠凝、增稠、乳化等流變學特性及其超分子結構之間的相互關聯,總結了影響多糖流變學特性的一般規律,旨在為多糖在食品工業中的進一步應用奠定理論依據。
1 多糖溶液特性
1.1 多糖稀溶液特性
當多糖完全水化后,以溶液形式存在。根據臨界接觸濃度c*的概念,多糖溶液一般可分為稀溶液和半稀溶液兩個區域。c*表示多糖分子在溶液中相互接觸的臨界濃度[16,31]。當多糖濃度低于c*時,多糖以單一分子存在,彼此不產生接觸和交聯;當濃度高于c*時,多糖分子開始發生彼此接觸甚至互相穿插、擠壓。因此,當多糖濃度越過c*時,溶液黏度特性將發生突變。通常,多糖溶液的c*和多糖分子在溶液中的流體力學體積有密切關系,流體力學體積越大,c*越小。由于特性黏度η是描述聚合物分子流體力學體積大小的度量,因此測定多糖溶液的η值可以估計流體力學體積。通常,不同類型的多糖分子質量、分支化程度以及分子構象均不同,因此它們的η值差異也較大。即便對于同一種多糖,當溶液所處的外界環境因素(如溫度、pH值、離子強度等)不同時,其分子構象不同,因此η值也不同。理論上,多糖的η值和c*具有反比關系,可以近似表示為c*=α/η,其中,α為常數,其值大小一般為4。顯然,這一經驗式為預測多糖溶液c*的簡單算法,但c*也可進行實際測定。
根據文獻[16]中不同質量分數(w)多糖溶液的增比黏度(ηsp)繪制圖1。lgηsp和lgw的關系可由兩個線性區表示:在多糖質量濃度較低時,斜率約為1.4;在多糖濃度較高時,lgηsp和lgw線性關系曲線的斜率約為3.3,兩個線性區的交點即為c*。當多糖濃度低于c*時,多糖分子彼此不接觸,此時多糖分子和水分子的摩擦力成為影響多糖溶液黏度的主要因素,當濃度高于c*時,多糖分子彼此接觸,產生較大的摩擦,此時多糖分子和水分子之間的摩擦、多糖分子彼此之間的摩擦共同決定溶液的黏度。因此,當多糖濃度高于c*時,溶液黏度明顯增大。然而,需要注意的是c*=α/η[32]這一經驗式僅適用于無規卷曲構象的多糖[32],即多糖分子彼此之間不發生聚集。但在一些情況中,多糖在溶液中常發生分子間聚集,導致實際測定的c*和采用經驗式推導的c*存在明顯偏差。這是因為當溶液中多糖分子彼此聚集形成分子間聚集體時,測定的η增大,由于c*和η為反比關系,使得實際測定的c*低于預測的c*。因此,通過對比預測的c*和實際測定的c*,可以間接推測出溶液中多糖 分子是否發生了聚集。即當實際測定的c*低于理論推測的c*時,表明發生了分子間聚集,反之則說明不存在分子間聚集[31]。
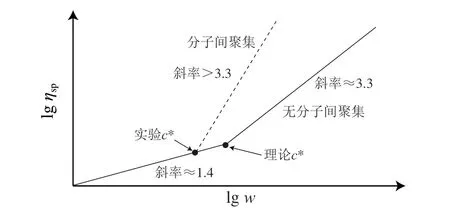
圖1 多糖溶液濃度與其增比黏度的關系[16]Fig.1 Relationship between the concentration of polysaccharide solution and its specific viscosity[16]
1.2 多糖濃溶液特性
當多糖濃度低于c*時,溶液中僅存在多糖分子和水分子之間的摩擦、水分子彼此之間的摩擦,兩者之和決定了溶液的黏度。通常,水分子之間的摩擦力很低,可忽略不計,因此多糖溶液的黏度主要取決于多糖分子和水分子之間的摩擦。此時,多糖溶液近似表現出牛頓流體的特性。當多糖的濃度高于c*時,多糖分子彼此接觸,導致多糖分子之間的摩擦力急劇增加,具體表現為溶液黏度顯著增加。此時,溶液的剪切稀化現象逐漸凸顯。由于不同結構特點的多糖在水溶液中的超分子結構不同,因此剪切稀化行為也不同,通常采用表觀黏度隨剪切速率的變化關系來描述多糖的剪切稀化效應。當剪切速率較低時,多糖分子聚集結構被外界剪切應力打破的速率和結構重建的速率幾乎相等,因此表觀黏度不隨剪切速率的增加而變化,此時溶液的特性黏度和零剪切黏度相等,可由η0表示。當剪切速率繼續增加時,多糖聚集結構的破壞速率大于重建速率,表觀黏度呈現降低趨勢,剪切速率越大,表觀黏度越低,這一階段即為剪切稀化區。當剪切速率繼續增加時,多糖聚集結構的破壞速率遠遠大于重建速率甚至多糖分子沿著剪切應力的方向發生定向重排,此時溶液的表觀黏度也不隨剪切速率的增加而增加,整體維持在較低的黏度水平(由η∞表示)。由于不同類型的多糖具有不同的分子結構,因此它們的流體力學體積不同,溶液中的超分子結構不同,因此它們的特性黏度變化趨勢也不同。
此外,當多糖濃度增加至c*以上時,多糖分子彼此接觸并互相擠壓,降低了單個多糖分子的流動性,因此當外界剪切應力打破溶液中多糖的超分子結構后,多糖分子總是需要一定的時間重新恢復聚集結構,濃度越大,多糖分子擠壓程度越高,恢復結構所需要的時間就越長,因此剪切稀化效應發生時所對應的剪切速率向更低的數值移動,根據文獻[32]數據繪制高濃度多糖溶液與低濃度多糖溶液剪切黏度變化如圖2所示。
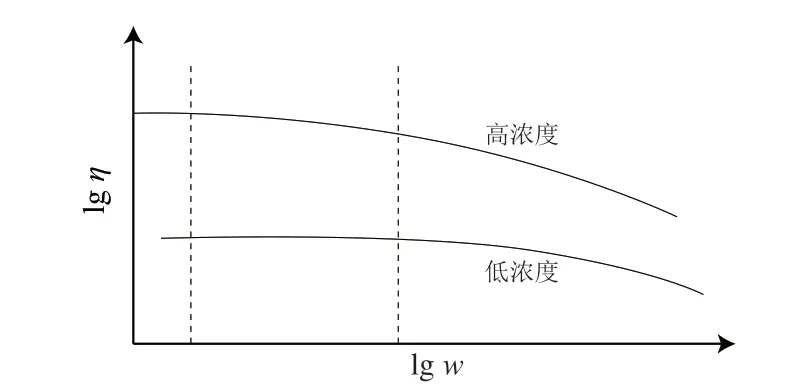
圖2 高濃度多糖溶液與低濃度多糖溶液剪切黏度變化[32]Fig.2 Shearing viscosity of polysaccharide solutions at high and low concentrations[32]
由于不同類型的多糖具有不同的η0,且同一類型的多糖在不同的濃度條件下也具有不同的η0,因此直接比較多糖溶液的黏度特征非常困難[33]。為了得到不同類型多糖剪切黏度的一般性規律,Morris等[32]定義多糖溶液特性黏度η降低至零剪切黏度η0十分之一時對應的剪切速率為“剪切稀化參數”,表示為γ0.1。如圖3所示,由不同濃度條件下瓜爾豆膠、L(lambda)型卡拉膠、刺槐豆膠、海藻酸鈉及透明質酸的歸一化結果可知,即便是多糖種類不同、分子質量不同、濃度不同,仍然可以獲得重合度較高的歸一化曲線,區別僅在于它們的η0和γ0.1不同。此外,Morris等[32]還發現,這一規律對溶液中發生明顯分子聚集行為的多糖也同樣適用。如圖3所示,刺槐豆膠、瓜爾豆膠、海藻酸鈉等均存在明顯的分子聚集行為,但仍然可以歸一化。原因可能是,雖然這些多糖在溶液中能夠發生分子聚集,但分子聚集所需要的時間遠遠超過多糖分子間彼此物理碰撞和接觸所需要的時間,因此在剪切作用下,分子聚集行為對溶液黏度的影響幾乎可以忽略不計。
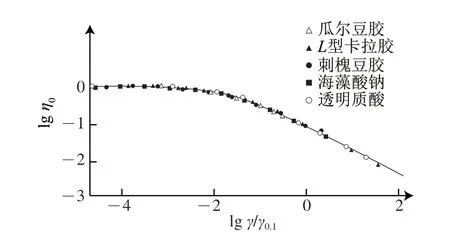
圖3 歸一化后多糖溶液黏度變化曲線[32]Fig.3 Generalized shear thinning behavior of concentrated solutions of disordered polysaccharides[32]
2 不同因素對多糖凝膠化的影響
當多糖超過某一臨界濃度時,有的多糖能在外界環境因素的誘導下發生凝膠化,稱為凝膠多糖,對應的臨界濃度稱為臨界膠凝濃度。然而,有關凝膠的定義卻一直沒有達成一致,主要原因在于評估多糖溶液體系是否形成凝膠的方法多種多樣,因此凝膠的定義也不盡相同。其中,最為簡單的方法是試管倒置法。當倒置試管時, 多糖體系不從試管底部流出,即可認為形成了凝膠。然而,流變學中,凝膠定義為儲能模量G′高于損耗模量G”的軟物質體系,只要符合這一標準,均可以從流變學角度判定為凝膠[34]。但某些凝膠體系由于結構較弱,雖然符合G′>G”,但并不符合試管倒置法的判別標準,通常將這樣的體系歸于弱凝膠的范疇。對于凝膠結構較強的多糖體系,G′應該比G”高一個數量級,且在較寬泛的振蕩頻率范圍內(如0.1~100 rad/s)G′和G”保持恒定,不隨振蕩頻率的增加而變化。目前,這一標準成為了食品流變學領域中評估食品多糖凝膠的主要依據,也排除了“弱凝膠”和“流體凝膠”的范疇。當多糖完全水化形成溶膠后,有的多糖體系能夠在改變外界環境因素的條件下形成凝膠,有的則不能。前者通常稱之為凝膠多糖,后者定義為非凝膠多糖。兩者最大的區別在于,凝膠多糖可以在水溶液中形成穩定的分子間交聯,構成空間三維網絡結構,但非凝膠多糖不能形成穩定的分子間交聯,因此在外界應力的作用下,非凝膠多糖的超分子結構很容易被打破,外觀表現為體系具有流動性。
圖4為兩種典型的多糖凝膠化機制[35]。對于凝膠多糖,凝膠化需要在合適的外界條件下發生,如適宜的溫度、pH值、離子濃度、多糖濃度等。對于負電荷密度較高的多糖,如海藻酸鈉和低酯果膠等,分子結構中含有較多數目的羧基,因此對二價離子(如Ca2+)具有較強的結合能力,通常一個鈣離子可以結合兩個解離的羧基基團[36-38]。當存在鈣離子時,海藻酸鈉和低酯果膠可以與鈣離子形成分子間交聯,構成穩定的空間三維網絡結構(圖4A)。這一凝膠化機制通常被描述為“蛋-盒模型”[39-42]。但對于某些食品多糖,它們本身具有良好的膠凝能力,可以在不添加鈣離子或其他陽離子的條件下形成凝膠。如圖4B所示,這類多糖往往電荷密度很低,凝膠化過程更多地依賴于分子螺旋聚集體的形成,主要包括結冷膠、瓊脂糖、卡拉膠等。然而,即便是對于這類凝膠多糖,添加陽離子也可以明顯促進凝膠化過程。一方面,添加陽離子屏蔽了多糖分子鏈段上的負電荷,降低了多糖分子間的靜電斥力,有利于螺旋聚集體的形成;另一方面,添加陽離子改變了水溶液的極性環境,降低了多糖的溶解度(鹽析效應),同樣也利于多糖分子彼此聚集。不同多糖凝膠的流變學特性如表1所示。
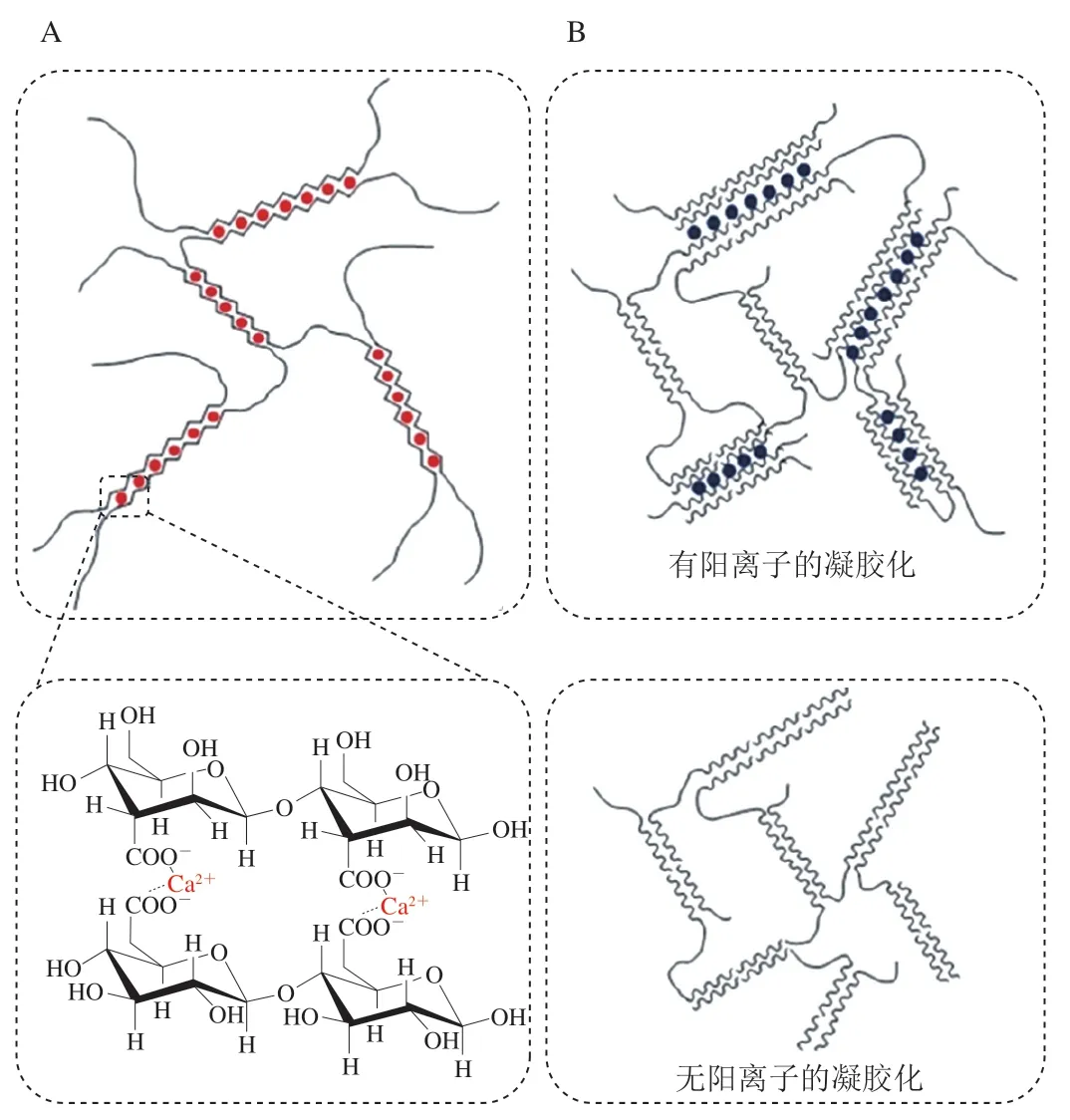
圖4 兩種典型的多糖凝膠化機制[35]Fig.4 Two typical gelling mechanisms of polysaccharides[35]

表1 不同多糖凝膠的流變學特性Table 1 Rheological properties of different polysaccharide gels
2.1 分子結構對多糖凝膠化的影響
即便是對于同一種多糖,分子結構的不同也會引起凝膠性質的較大差異。一般而言,多糖分子質量越大,越有利于形成凝膠。此外,對于海藻酸鈉和低酯果膠這類依靠鈣離子存在而發生凝膠化的多糖,分子鏈段上羧基的分布模式也是影響與鈣離子結合的重要因素。通常, 當羧基隨機分布于多糖鏈段時,膠凝能力較弱,當羧基連續分布于多糖鏈段時(“區域”分布模式),更有利于形成凝膠。有研究表明,當連續6 個解離的羧基和鈣離子結合時,相鄰的果膠分子才能形成穩定的交聯區,顯然,“區域”分布模式更利于多糖分子與鈣離子結合形成穩定交聯區[3]。多糖分支化程度也對多糖的膠凝能力有一定影響。在同等分子質量的前提下,分支化程度越高的多糖,流體力學體積越低,因此不利于形成濃溶液,一定程度上降低了多糖的膠凝能力。尤其是對于低酯果膠而言,分支化程度越高,分子間空間位阻越大,越影響果膠分子“蛋-盒”模型的形成。然而,近年來也有報道指出,分支化程度較高的果膠具有更好的膠凝能力,這是因為側鏈分支相互纏繞,有助于凝膠網絡的形成。
此外,某些關鍵基團的含量也會對多糖的凝膠化過程和凝膠特性產生明顯影響,例如果膠、結冷膠及卡拉膠等。高酯果膠和低酯果膠最主要的區別是酯化程度不同,但足以使兩者具有不同的膠凝機制[3]。低酯果膠凝膠化需要鈣離子參與,凝膠可以在相對較寬的pH值范圍(如pH 3.5~9.5)內形成[36],但高酯果膠凝膠化不需要鈣離子參與。結冷膠分為高酰基和去酰基兩種形式,微生物發酵后直接產生的結冷膠為高酰基形式,通過熱堿處理除去高酰基結冷膠分子鏈上的酰基基團后可得到去酰基結冷膠[50]。去酰基結冷膠的膠凝機制一般解釋為:高溫條件下,結冷膠以無規卷曲構象存在,隨溶液溫度降低,結冷膠分子逐漸形成雙螺旋結構,當溫度進一步降低時,雙螺旋彼此聚集形成螺旋聚集體,引起空間三維網絡形成,導致凝膠化[51]。加熱時,結冷膠凝膠逐漸融化,但凝膠融化溫度通常高于60 ℃,呈現明顯的熱滯后效應。高酰基結冷膠的酰基取代基分為乙酰基和甘油酰基兩種,據報道顯示,乙酰基被認為是阻礙高酰基結冷膠分子螺旋聚集體形成的關鍵因素,而甘油酰基主要起到穩定高酰基結冷膠雙螺旋的作用[52]。因此,高酰基結冷膠凝膠形成過程和凝膠融化過程中均不涉及螺旋聚集體的形成和解螺旋,所以高酰基結冷膠沒有明顯的熱滯后效應,凝膠化溫度和凝膠融化溫度幾乎一致[53]。類似的例子還包括卡拉膠,卡拉膠主要包括K型(kappa)、I型(iota)、L型3 種形式,區別僅在于硫酸酯基含量和連接位點不同,但足以引起它們在膠凝特性方面的差異[54-58]。例如,K型卡拉膠對鉀離子非常敏感,但對鈉離子不敏感,在K+存在的前提下,K型卡拉膠的凝膠化過程被極大促進,形成強度較高、質地硬而脆的凝膠。L型卡拉膠對一價陽離子和二價陽離子均不敏感。I型卡拉膠可以和鈣離子結合形成柔軟富有彈性的凝膠。
2.2 外界因素對多糖凝膠化的影響
凝膠化的本質是多糖水溶液在適當的外界刺激下,多糖分子發生聚集,形成穩定的分子間聚集區,從而形成 三維網絡結構的過程。通常,對于冷致型凝膠多糖,降低溫度可以促使多糖溶液從溶膠態轉變為凝膠態。對于疏水基團含量較高的多糖,疏水作用力是誘導發生凝膠化的主要因素[2]。當加熱這類多糖的水溶液至凝膠化溫度以上時,凝膠才能形成,稱之為熱致凝膠。例如,甲基纖維素(methylcellulose,MC)和羥丙基甲基纖維素(hydroxypropyl methyl cellulose,HPMC)均是典型的熱致型凝膠多糖。有報道顯示,MC的凝膠化溫度約為52 ℃,HPMC的凝膠化溫度范圍較寬,約為63~80 ℃[2]。當冷卻MC或HPMC的凝膠時,分子間疏水作用力逐漸減弱,導致凝膠網絡解聚集,促使多糖分子從凝膠狀態重新轉變為溶液狀態。pH值對多糖凝膠化的影響在于改變帶電荷多糖的分子構象以及分子間相互作用力。因此,改變溶液pH值可以誘導一些多糖發生凝膠化,或促進凝膠形成。例如,據報道顯示,結冷膠溶液的pH值降低至3.5時,凝膠強度可增加4 倍以上[59]。此外,pH值也是影響海藻酸鈉凝膠化的重要因素,海藻酸鈉電荷密度較高,在不含二價陽離子的情況下,溶液一般以黏稠流體的形式而存在。然而,當pH值緩慢降低至3.0時,海藻酸鈉可形成凝膠,原因在于降低溶液pH值后,減少了海藻酸鈉分子間的靜電斥力,有利于分子聚集。陽離子對多糖凝膠化的影響分為3 個方面:1)一價陽離子通過遮蔽多糖分子鏈上的負電荷降低分子間靜電斥力,促進分子間聚集;2)二價或多價陽離子直接和多糖分子中解離的羧基結合形成分子間交聯;3)陽離子濃度過高時,改變了溶液的極性環境,可能引起多糖分支的“鹽析”效應。
此外,多糖的凝膠化過程也受其他因素的影響。例如,在少量蔗糖存在的情況下,低酯果膠的凝膠強度明顯提升,一方面,蔗糖作為脫水劑,破壞了果膠分子表面的水化膜,有利于果膠分子彼此靠近與鈣離子形成交聯;另一方面,蔗糖本身也可以作為填充成分加強果膠分子的凝膠網絡。
3 非凝膠多糖
與凝膠多糖相比,非凝膠多糖水化后以流體形式存在,因此在食品領域中主要用作增稠劑、分散劑、乳化劑等。需要注意的是,所有的凝膠多糖均具有增稠特性,這是由它們的高分子本質決定的。例如,當凝膠多糖在水溶液中的濃度較低,不足以形成較強的三維凝膠網絡時,溶液宏觀上表現為黏稠體系,和非凝膠多糖相似。某些多糖,如黃原膠、瓜爾豆膠、魔芋膠等,自身可以形成微弱的分子間聚集,使得它們的溶液常表現出類似于弱凝膠的特點,即G′>G”,但這樣的結構容易被剪切應力破壞而產生流動性。因此,這類多糖仍然歸屬于非凝膠多糖。
食品工業中,非凝膠多糖主要用作增稠劑。由于不同結構特點的多糖在溶液中的超分子結構不同,因此溶液黏度特征也不同。除上述總結的多糖溶液特性外,剪切稀化行為是評估非凝膠多糖流變特性的另一重要因素,通常采用表觀黏度隨剪切速率的變化關系來描述。通常,不同結構特點的多糖剪切稀化行為呈現較大差異。例如,黃原膠一般具有最為顯著的剪切稀化效應,低剪切速率條件下,黃原膠溶液表觀黏度很高,隨剪切速率增加,表觀黏度急劇降低,使得黃原膠具有最為明顯的剪切稀化效應。其他具有顯著剪切稀化效應的多糖主要為葡甘露聚糖類。主要原因是,黃原膠和葡甘露聚糖均能發生微弱的分子間聚集,形成類似于“弱凝膠”的結構,因此低剪切速率時,溶液表觀黏度很高,但這種結構容易被剪切應力破壞,所以高剪切速率時,溶液黏度急劇降低[60-63]。具有顯著剪切稀化效應的非凝膠多糖一般作為性能優良的增稠劑或穩定劑廣泛應用于飲料工業中。
同樣地,非凝膠多糖的增稠特性也受到外界環境因素,如溫度、pH值、鹽離子等的影響。一方面,這些因素可以影響非凝膠多糖的分子構象,進一步影響多糖的流體力學體積;另一方面,這些因素也可以改變多糖分子間作用力,促進多糖分子聚集,形成微弱的聚集網絡,增加溶液黏度。例如,對于含有羧基的非凝膠多糖,隨pH值增加,多糖分子的電荷密度也增加,分子間靜電斥力增大,多糖分子擴張,流體力學體積增大,有利于增加溶液黏度。然而,加入陽離子時,多糖分子攜帶的負電荷被屏蔽,降低了分子間靜電斥力。但在某些特殊情況中,當多糖分子攜帶的電荷被屏蔽后,多糖可能發生分子間聚集,形成微弱的三維網絡結構,反而增加了多糖溶液黏度。當溫度升高時,對于以分子間氫鍵為主要作用力的多糖,其溶液黏度往往呈現出降低趨勢,但對于以疏水作用為主要結合力的多糖而言,溶液黏度往往表現出明顯的增加,甚至導致凝膠化。
需要注意的是,非凝膠多糖和凝膠多糖的區分并沒有嚴格的界限,而是根據多糖在食品工業的常規用途而進行的劃分。在一些特殊的條件下,某些非凝膠多糖也可能發生凝膠化。例如,有報道顯示,黃原膠在凍融條件下可以發生凝膠化[53]。同樣地,當嚴格控制使用條件時,某些凝膠多糖也可能不發生凝膠化。
4 表面活性多糖
4.1 表面活性多糖的界面吸附特性
區別于小分子表面活性劑,表面活性多糖的表面活性特指多糖分子吸附在油-水界面的能力,而小分子表面活性劑的表面活性則指降低油-水界面張力的特性。多糖的 表面活性構成了其作為乳化劑的基礎[21]。通常,多糖乳化劑降低油-水界面張力的能力非常有限,因此多糖的乳化能力主要來源于兩個方面:1)吸附在油-水界面的能力;2)在乳滴表面形成一層水化膜,防止油滴聚集的能力[19]。一旦乳液形成,需要多糖分子永久性地吸附在油-水界面,提供保護作用,這就要求多糖分子必須含有一定數目的疏水基團[64-66]。此外,乳滴表面水化膜的厚度也是影響多糖乳液穩定性重要因素,水化層越厚,乳滴之間的空間位阻作用也越大,乳液穩定性越好。除疏水基團含量外,影響多糖乳液穩定性的因素還包括多糖的電荷密度、分子質量,以及乳液離子強度、溫度等[67-68]。近年來,國內外對多糖乳化特性的研究越來越重視,相關報道日益增多,目前國內已有不少文獻詳細總結了多糖的乳化特性及多糖乳液的穩定策略[69-75]。
4.2 表面活性多糖的分子聚集行為
表面活性多糖的另一個特殊性質是其在水溶液中的分子聚集特性[76-77]。表面活性多糖分子結構中含有一定數目的疏水基團,因此疏水作用力可能會對多糖在溶液中的存在狀態造成一定影響。如果疏水作用力足夠強烈,多糖分子能夠在疏水作用力的作用下形成以疏水微區或膠束為交聯的分子間聚集結構[78]。一般地,當表面活性多糖濃度增加到某一臨界值時,多糖水溶液中開始形成膠束,這一濃度通常稱為臨界膠束濃度。多糖的臨界膠束濃度概念和小分子表面活性劑的臨界膠束濃度概念類似,但由于多糖溶液同時又兼具聚合物溶液的一般特性,使得表面活性多糖的分子聚集行為更加復雜[79]。此外,表面活性多糖屬于聚合物表面活性劑的范疇,而聚合物表面活性劑在水溶液中的聚集行為受疏水基團含量和分布模式的影響。有文獻報道,根據聚合物表面活性劑疏水官能團的分布模式,可將聚合物表面活性劑分為大分子表面活性劑和聚皂兩大類[72]。大分子表面活性劑的結構和小分子表面活性劑結構類似,是由親水頭鏈和疏水尾鏈共同構成的聚合物分子。聚皂則指疏水單元和親水單元嵌段形成的共聚物分子。因此,根據定義來看,幾乎所有的天然表面活性多糖和疏水改性后的多糖均屬于聚皂。然而,大多數天然多糖所含的疏水基團數目有限,因此形成膠束的能力較弱。目前只有零星研究報道了天然表面活性多糖形成膠束或疏水微區的能力。例如,有研究報道,高酰基結冷膠凝膠中存在明顯的疏水微區,且隨多糖濃度降低,疏水微區的密度也降低[31]。
5 結 語
多糖種類繁多,結構差別較大,不同的多糖常表現出各異的流變學特性。外界環境因素影響多糖流變學特性的本質在于影響多糖的超分子結構,因此多糖分子 在溶液中的聚集結構是影響多糖流變學特性的根本因素。本文通過綜述多糖流變學特性的基本原理,可為多糖在食品工業中的應用提供理論參考。
