鐵死亡在急性腎損傷和腎癌等相關疾病中的研究進展
2021-08-18 09:08:54葉承林綜述劉修恒審校
疑難病雜志 2021年8期
葉承林綜述 劉修恒審校
細胞死亡有多種形式,在20世紀60年代,人們認識到細胞死亡可以通過分子機制來調控,并且可以為正常生理功能提供幫助,也能導致病理變化,“程序性細胞死亡”這一概念隨之被提出。細胞死亡曾被分為三大類,Ⅰ型細胞死亡即細胞凋亡;Ⅱ型細胞死亡又叫自噬相關性細胞死亡;Ⅲ型細胞死亡被稱為細胞壞死[1]。最近的研究發現,調節性細胞死亡有多種形式,其分子機制和形態特征各不相同。鐵死亡(ferroptosis)這一概念于2012年首次提出,是一種鐵依賴的非凋亡性細胞死亡。這種細胞死亡機制不需要含半胱氨酸的天冬氨酸蛋白水解酶(caspase)的激活,也不需要其他凋亡效應分子(如BAX或BAK)的參與,也不伴隨凋亡的形態特征或生化過程。形態學上主要表現為線粒體的縮小、線粒體嵴減少或消失[2]。研究證明鐵死亡在腫瘤、缺血再灌注、神經退行性病變等領域發揮重要作用,其在腎臟相關疾病中的重要性也逐漸被發現,本文就鐵死亡的發現、作用機制與調控及在腎臟疾病發生發展中的作用展開綜述,為腎臟疾病的進一步研究提供新的方向。
1 鐵死亡概念
在鐵死亡的概念被提出及詳細分子機制發現之前,已經有很多文獻報道了對于這一現象的觀察,但都被歸因于其他的細胞死亡機制,或者被認為沒有生物學意義。20世紀50年代中期Eagle等[3-5]發現13種不同氨基酸中只有一種氨基酸饑餓抑制了人和小鼠細胞的生長;被剝奪胱氨酸的細胞表現出獨特的顯微形態,與被剝奪其他氨基酸時的形態不同,推測與病毒感染引起的細胞死亡相似。1977年Bannai等[6]發現胱氨酸饑餓導致細胞谷胱甘肽減少和細胞死亡,通過添加親脂性抗氧化劑α-生育酚(α-tocopherol,維生素E的一種成分)可以挽救這種胱氨酸剝奪所致的死亡,從而支持了活性氧(ROS)積累在細胞死亡誘導中的貢獻,并且這一過程無需谷胱甘肽參與。Ratan等[7]發現由于剝奪血清而導致的鐵缺乏可以阻止細胞胱氨酸剝奪和谷胱甘肽耗竭在誘導細胞死亡中的關鍵作用,現在被認為是鐵死亡的特征表現之一。
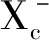
Dixon等[11]于2012年發現首個鐵死亡抑制劑ferrostatin-1,并且在這些研究基礎上正式提出了鐵死亡(ferroptosis)這一概念,用以描述這種鐵依賴性非凋亡性的細胞死亡。
2 鐵死亡機制與調控
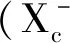
2.2 脂質過氧化與活性氧(ROS)的蓄積 ROS通常被認為是氧消耗和細胞代謝的副產品,由分子氧的部分還原形成[21]。腫瘤內源性活性氧主要來源于細胞線粒體及NADPH氧化酶(NOXs)[22]。過量的ROS被抗氧化劑通過酶促或非酶促反應及在超氧化物歧化酶(superoxide dismutase,SOD)如Cu/Zn-SOD、Mn-SOD等,谷胱甘肽過氧化物酶(glutathione peroxidase,GPX)和過氧化氫酶催化的反應中解毒。ROS產生和解毒速率的不平衡會導致氧化應激,并由此產生自由基,這些自由基會破壞DNA、蛋白質和脂質。氧化還原活性金屬,特別是鐵,可以通過芬頓反應促進細胞中的ROS聚集。在鐵死亡過程中,胱氨酸—谷氨酸逆向轉運蛋白(recombinant solute carrier family 7,member 11,SLC7A11)的失活和谷胱甘肽的耗竭導致鐵依賴的ROS積聚[15]。
多不飽和脂肪酸(polyunsaturated fatty acids,PUFAs)的過氧化是鐵死亡的主要驅動因素。鐵依賴的脂質活性氧的積累和隨后多不飽和脂肪酸磷脂(PUFA-PLS)的消耗是鐵死亡的共同特征[23]。花生四烯酸(arachidonic acid,AA)是目前已知的鐵死亡細胞中最常消耗的多不飽和脂肪酸[24]。Dixon等[25]的研究發現,將花生四烯酸插入膜磷脂中所涉及的關鍵酶缺失可以防止鐵死亡的發生。酰基輔酶A合成酶長鏈家族成員4(acyl-CoA synthetase long chain family member 4,ACSL4)可通過促進可氧化的細胞膜磷脂的積累來驅動鐵死亡[26]。
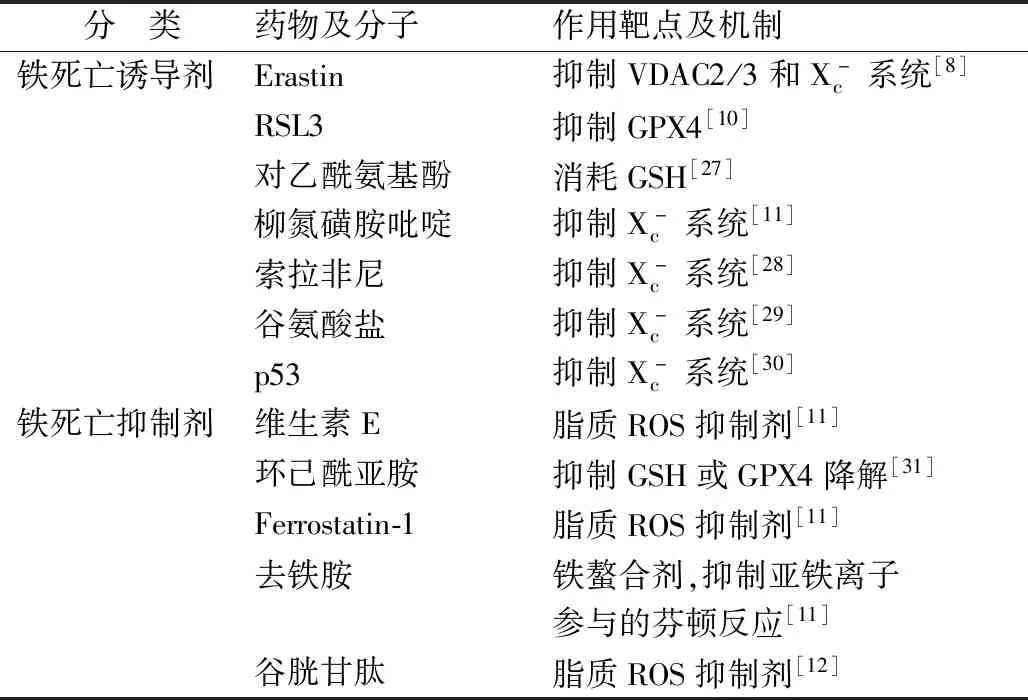
表1 鐵死亡調控相關誘導劑與抑制劑
3 鐵死亡與急性腎損傷
急性腎損傷(acute kidney injury,AKI),也稱為急性腎衰竭(acute renal failure,ARF),是由多種原因引起的常見嚴重疾病,病因包括腎缺血、腎毒性藥物和尿路阻塞[32]。AKI的發病機制非常復雜,由于其解剖學特征和復雜功能,腎單位的近端腎小管節段最容易受到各種形式的損傷[33]。已經發現了多種分子機制可以誘導或加重AKI,其中ROS被認為是誘導腎損害的關鍵介質之一[34]。多項研究表明,鐵死亡是潛在的治療靶點,尤其是在腎小管壞死占主導的疾病中[35]。
在腎臟中,缺血再灌注損傷(ischemia-reperfusion injury,IRI)是AKI的主要原因之一。IRI的特征是特定器官的血液供應突然停止并在血流恢復后重新供氧。該過程可通過觸發涉及ROS、細胞因子、趨化因子和白細胞激活的炎性級聯反應來加劇組織損傷[36-37]。以往在各種缺血性損傷模型中,凋亡被認為是主要的調控性細胞死亡。當壞死性凋亡被發現時,這種受體相互作用的絲氨酸/蘇氨酸激酶3(receptor interacting serine/threonine kinase 3,RIPK3)和混合譜系激酶結構域樣(mixed lineage kinase domain like,MLKL)依賴性調節性壞死的形式被認為是心臟及腎臟缺血性損傷的主要原因[38-39]。最近的發現表明,鐵死亡可能是缺血性損傷的主要驅動力之一。在嚴重的IRI模型中應用鐵抑素(ferrostatin)可保護小鼠免受功能性急性腎衰竭,然而RIPK1抑制劑necrostatin-1(Nec-1)不能保護新鮮分離的腎小管免受缺氧損傷[40],這突顯了鐵穩態在人類缺血再灌注損傷中的重要性。基礎研究證明,NRF2過度激活會增加GSH的產生并阻止腎臟IRI早期階段的進展[41],許多負責阻止脂質過氧化進而促進鐵死亡的蛋白質和酶都是NRF2的靶基因,如SLC40A1、谷胱甘肽合成酶、GPX4。NRF2或其靶基因的功能失調與對細胞應激源的反應性下降有關,并誘導細胞鐵死亡[42]。Lee等[43]研究表明,用能量應激誘導劑AICAR或2-Deoxy-D-glucose(2DG)處理可以通過抑制鐵死亡保護小鼠免受腎臟IRI的影響。
橫紋肌溶解引起的腎衰竭占所有急性腎衰竭病例的15%[44],研究表明,肌紅蛋白(myoglobin,Mb)在腎臟中的積累是導致腎臟損害的核心機制。近年來,對橫紋肌溶解誘導的急性腎損傷研究表明,Mb代謝產生的Fe2+直接誘導近端腎小管上皮細胞脂質過氧化可能是橫紋肌溶解致腎損傷的重要機制之一[45]。腎臟中Mb降解釋放的游離鐵通過芬頓反應的催化作用參與氧化性物質的產生。研究表明,使用鐵絡合劑去鐵胺可以減輕橫紋肌溶解誘導的大鼠腎臟損傷[46]。Guerrero-Hue等[47]證明了鐵死亡在橫紋肌溶解誘導AKI中的關鍵作用,并表明鐵死亡敏感的這一過程可以被強抗氧化劑姜黃素所抑制。
4 鐵死亡與腎癌
腎細胞癌根據病理特征可分為腎透明細胞癌、乳頭狀腎細胞癌、嫌色細胞性腎細胞癌及Bellini集合管癌等,其中以腎透明細胞癌最為常見。腎癌多對化療和放療耐藥。激活調控性細胞死亡是腎癌的理想治療策略,并可能有助于解決腎癌的耐藥性。
鐵死亡首先在非小細胞肺癌HT-1080細胞系中被鑒定[11],隨后,Yang等[12]測試了117個腫瘤細胞系對erastin引起鐵死亡的敏感性,并發現彌漫大B細胞淋巴瘤和腎細胞癌對GPX4調節的鐵死亡尤為敏感。Miess等[48]發現siRNA沉默谷胱甘肽過氧化物酶GPX3和GPX4對腎透明細胞癌細胞是致命的,并且闡述了通過抑制GSH的合成,腎透明細胞癌對于鐵死亡變得更加敏感,從而抑制腫瘤的生長。在缺乏VHL基因的腎癌細胞中,重新表達的VHL產生了對鐵死亡的抵抗力。最近的一項研究表明,能量應激(energy stress)介導的AMPK激活能夠抑制鐵死亡,在腎癌Caki-1細胞系(一種基礎AMPK活性較低的鐵死亡敏感細胞系)中過表達 AMPK可適度增加乙酰輔酶A羧化酶(acetyl CoA carboxylase,ACC)的磷酸化,并保護細胞免受Erastin誘導的鐵死亡影響[43]。Yang等[49]的一項研究表明,Hippo通路效應器TAZ可以調節腎癌對鐵死亡的敏感性。Kerins等[50]發現,延胡索酸水合酶的失活可以導致細胞內延胡索酸的蓄積從而提高遺傳性平滑肌瘤病和腎細胞癌(hereditary leiomyomatosis and renal cell cancer,HLRCC)對鐵死亡的敏感性。
5 鐵死亡相關研究的展望
鐵死亡作為一種調控性的細胞死亡,在腎臟疾病中具有雙重作用,對于鐵死亡在腎臟疾病中的有關機制仍需要進一步探索。鐵死亡機制層面上,盡管已經發現了脂質過氧化物在誘導鐵死亡中的重要作用,但仍然沒有確鑿的證據表明此類ROS是執行鐵死亡的最下游因素,關于鐵死亡的最終執行分子問題仍有待解決。鐵死亡與其他調控性細胞死亡如自噬、凋亡之間的聯系需要進一步深入研究。鐵死亡作為細胞調控性死亡的一種形式,在其作用過程中不可逆點的位置,正在進行鐵死亡這一過程的細胞在何時可以被挽救,這些都需要深入的研究。鐵死亡是腎臟疾病中非常重要的細胞死亡形式之一,隨著相關研究的不斷進展,一定會為腎臟疾病的治療帶來新的方向。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
四川勞動保障(2021年9期)2022-01-18 05:11:08
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
文苑(2018年21期)2018-11-09 01:23:06
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國衛生(2016年9期)2016-11-12 13:28:08
中國衛生(2015年9期)2015-11-10 03:11:12
