高溫花生粕蛋白基膠黏劑的制備與表征*
2021-08-09 11:42:08趙紫蕓岳海濤
林業科學 2021年6期
關鍵詞:改性
屈 陽 郭 芹 李 甜 趙紫蕓 岳海濤 楊 潔 王 強
(1.新疆大學生命科學與技術學院 烏魯木齊 830046;2.中國農業科學院農產品加工研究所 農業部農產品加工與質量控制重點開放實驗室 北京 100193)
我國是世界上最大的木制品生產國、加工國和貿易國,2018年人造板供給量為2.99億m3,居世界第一,其木材膠黏劑消費量達1 877萬t,主要為依賴石油資源的“三醛膠”(脲醛樹脂、酚醛樹脂和三聚氰胺甲醛樹脂)(顧繼友,2015)。“三醛膠”合成時主要原料之一是甲醛,反應過程中存在未反應完全的甲醛,使用過程中受光照、高溫、氧等作用也會緩慢釋放甲醛,所以只要使用“三醛膠”就不能從根本上解決甲醛污染問題。近年來,隨著石油價格的攀升以及人們環保意識的提高,綠色無污染的生物質膠黏劑(單寧、木質素、淀粉、植物蛋白、動物蛋白等)(龐艷芳等,2018;Heinrich,2019)逐漸成為人們關注的焦點,但由于傳統的生物質膠黏劑耐水性差、膠合強度低,在實際應用中受到限制。采用改性、交聯、共聚等方法可有效提高生物質膠黏劑的耐水性和膠合強度(Lietal.,2012;2015a;2018),現有研究多集中于大豆蛋白基膠黏劑(Zhaoetal.,2018;Qietal.,2016;Liuetal.,2015;Changetal.,2017),其他生物質膠黏劑的研究報道較少。
高溫花生粕(hot-press peanut meal,HPM)是花生油生產的主要副產品,我國高溫花生粕年產量達400萬t,但由于2017年國家頒布的《飼料衛生標準》(GB 13078—2017)限定黃曲霉毒素B1 ≤50 μg·kg-1,使得大量高溫花生粕不能用于飼料行業而造成浪費。花生粕價格低廉、來源廣泛,粗蛋白含量高達40%~50%,其中球蛋白含量約85%(李潤嬌,2014),是加工生物質膠黏劑的良好蛋白原料。Yang等(2006)最早使用花生粕、大豆粕和動物血清蛋白與酚醛樹脂共混制備木材膠黏劑,但其耐水性差。Li等(2015b)在此基礎上采用十二烷基硫酸鈉(SDS)和乙二醇縮水甘油醚(EGDE)改性制備高溫花生粕蛋白基膠黏劑,耐水性和強度顯著提高。實驗室前期研究(Chenetal.,2018)發現,高溫花生粕比低溫花生粕更適合制備蛋白基膠黏劑,且制備的膠黏劑完全滿足國家Ⅱ類楊木膠合板使用標準。采用高溫花生粕制備蛋白基膠黏劑不僅能解決高溫花生粕難以高值化利用的問題,而且符合當前綠色無醛的環保理念,具有較好的應用前景。
鑒于此,本研究以高溫花生粕(HPM)為試驗材料,采用SDS、納米SiO2(nSiO2)和聚酰胺多胺環氧氯丙烷(PAE)樹脂復合改性交聯制備符合國家Ⅰ類膠合板要求(室外使用)的HPM蛋白基膠黏劑(HPMA),并分析改性過程中其性能和結構的變化,以期為HPMA的產業化應用提供技術和理論依據。
1 材料與方法
1.1 試驗材料與儀器
1.1.1 試驗材料 高溫花生粕,工業級(蛋白含量48%),山東玉皇糧油有限公司;PAE,工業級,浙江傳化集團有限公司;楊木單板(幅面250 mm×250 mm×1.6 mm),市售,山東臨沂有你之家板材公司;SDS,分析純,北京索萊寶科技有限公司;nSiO2,分析純,上海麥克林生化科技有限公司。
1.1.2 試驗儀器 CARVER25-12H型熱壓機(300 mm×300 mm),美國邁可諾技術有限公司;WDW-20E微機控制電子萬能試驗機,濟南時代試金試驗機有限公司;PB-10型pH計,賽多利斯科學儀器北京有限公司;SQW-100DF超微粉碎機,濟南易辰超微粉碎技術有限公司;TENSOR27傅里葉變換紅外光譜儀,BURUKER公司;Pyris Diamond熱重-差熱綜合熱分析儀,美國Perkin Elmer公司;SU8010掃描電子顯微鏡,日本Hitachi公司。
1.2 試驗方法
1.2.1 蛋白基膠黏劑制備 稱取180 g水于燒杯中,升溫至60 ℃后加入60 g高溫花生粕粉,攪拌30 min(取樣,標記為A),然后加入1.92 g SDS攪拌30 min(取樣,標記為B),再添加1.2 g nSiO2攪拌30 min(取樣,標記為C),最后加入51 g PAE交聯30 min,制得改性高溫花生粕蛋白基膠黏劑(取樣,標記為D,即HPMA)。
1.2.2 膠合板制備 壓制3層楊木膠合板。采用手工涂膠方式將膠黏劑均勻涂抹在楊木單板上,單面施膠量220 g·m-2,涂膠完成后閉合陳放10 min進行熱壓處理,熱壓溫度120 ℃,熱壓時間85 s·mm-1,熱壓壓力1.2 MPa。
1.2.3 膠合板膠合強度分析 按照GB/T 9846—2015剪裁膠合板試件,在63 ℃水中浸泡3 h,室溫冷卻10 min后使用微機控制電子萬能試驗機檢測其溫水濕態膠合強度;將裁好的膠合板試件在沸水中浸泡4 h,然后置于鼓風干燥箱內60 ℃干燥20 h,再將其在沸水中浸泡4 h,最后在不超過30 ℃的冷水中浸泡1 h,室溫冷卻10 min后使用微機控制電子萬能試驗機檢測其沸水濕態膠合強度。
1.2.4 蛋白基膠黏劑的物化性能分析 外觀、黏度、固體含量按照GB/T 14074—2006測定。
1.2.5 蛋白基膠黏劑的熱穩定性 精確稱取5 mg樣品,通入氮氣條件下以20 ℃·min-1加熱,TG/DTA分析樣品在30~550 ℃的質量變化。
1.2.6 蛋白基膠黏劑的紅外光譜分析 將試樣冷凍、干燥、固化后研磨成粉,與溴化鉀混合,壓片后采用傅里葉變換紅外光譜儀進行全波段掃描。
1.2.7 蛋白基膠黏劑的微觀形態分析 樣品經冷凍、干燥后自然斷裂,用鑷子將斷裂面朝上固定好,并進行噴金處理,采用掃描電子顯微鏡分析10 kV加速電壓下蛋白基膠黏劑的微觀形態。
1.2.8 數據分析 使用Origin 8.5軟件作圖,采用SPSS公司SPSS 22.0軟件的Duncan檢驗進行顯著性分析,取α=0.05,重復3次(n=3)。各項指標的測定值以平均值±標準誤表示。
2 結果與討論
2.1 HPMA制備過程的膠合強度分析
由表1可知,復合改性交聯對蛋白基膠黏劑的膠合強度影響較大。A的膠合強度最低,耐水性差,膠合板在測試過程中均開裂。加入SDS改性后制得B的干態膠合強度提高58%,可能是因為SDS破壞了蛋白質分子內的二硫鍵和氫鍵,使蛋白質二、三級結構舒展并暴露內部疏水基團形成疏水膠束,其耐水性和強度提高(Huangetal.,2000;Wangetal.,2005);B的溫水濕態膠合強度高于國家Ⅱ類楊木膠合板標準(0.70 MPa),但其沸水濕態膠合強度較低。加入nSiO2制得C的干態和溫水濕態膠合強度有所提高,可能是因為nSiO2粒徑小,填充在蛋白基膠黏劑作用的木材空隙中使連接更牢固(張學軍,2008),但其沸水濕態膠合強度仍然低于0.70 MPa,不滿足國家Ⅰ類膠合板標準。D(HPMA)與李婧婧(2016)制備的改性花生蛋白基膠黏劑相比,溫水濕態膠合強度提高27%,且更耐沸水,滿足國家Ⅰ類膠合板標準,既適宜于室內使用,也適宜于室外使用。D的膠合強度顯著提高可能是因為PAE與花生蛋白發生交聯反應生成超支化的網絡結構(蔣瑞等,2017;Lietal.,2004),同時nSiO2能夠均勻填充到交聯網絡中,形成立體網絡銀紋結構,因此進一步提高了膠合強度和延展性。
2.2 HPMA制備過程的物化性能分析
復合改性交聯對HPMA外觀顏色影響不顯著,A、B、C、D均為棕褐色不透明液體(表2),主要由原料本身顏色決定。黏度是評價木材膠黏劑的重要指標,過高或過低均會影響膠合強度,對于軟木材和干木材來說最適宜的黏度為5 000~25 000 mPa·s(陳云等,2014)。由表2可知,A的黏度過低,B的黏度與A相比增加72.98%,可能是SDS展開蛋白質二、三級結構后使蛋白分子之間軸距減小,摩擦增多,因此黏度提高。C的黏度與B相比顯著增加389.19%,可能是加入nSiO2后,蛋白質分子氨基、羧基與nSiO2形成分子內氫鍵,阻止膠黏劑流動,提高了固化速率,膠黏劑黏度急劇升高,但仍然滿足適宜黏度范圍。D與C相比黏度下降37.80%,其涂抹性適宜,且制備的膠合板耐沸水。
由表2還可知,A的pH呈弱酸性,加入SDS后B的pH升高為弱堿性,加入nSiO2后C的pH變化不明顯,加入PAE后D的pH顯著降低,呈弱酸性,這主要是因為PAE的pH呈酸性(5.5),降低了整個體系的pH。此pH與花生蛋白的等電點(pH=5.0)接近(徐飛等,2016),有利于提高HPMA的膠合強度。復合改性交聯過程中,HPMA的固體含量逐漸增加,由19.6%升至31.33%,且在正常的蛋白基膠黏劑固體含量范圍(20%~35%),在此范圍內,固體含量增加既可減少熱壓過程中的起泡現象,還能提高膠合強度(陳焱,2018)。以上結果表明,制備的HPMA物化性能較好。

表2 HPMA制備過程的物化性能①Tab.2 Physico-chemical properties of HPMA preparation process
2.3 HPMA制備過程的熱穩定性分析
HPMA制備過程的TG和DTG曲線如圖1所示。由TG曲線可知,A、B、C、D變化趨勢基本一致,質量損失主要包括3個階段:25~200 ℃,質量下降較少,可能是殘存水分的蒸發(Liuetal.,2015);200~360 ℃,質量損失較大,可能是蛋白質在此階段發生降解;360~550 ℃,質量也會損失,可能是該溫度下一些化學鍵會發生斷裂,生成CO、H2S等氣體,與Li等(2015b)研究結論基本一致。由DTG曲線可知,A的熱分解溫度為299.9 ℃,B的熱分解溫度提高13.8 ℃,加入nSiO2后C的熱分解溫度變化不明顯(314.8 ℃),加入PAE后D的熱分解溫度與A、B、C相比達到最高(317 ℃),說明復合改性交聯可提高膠黏劑的耐熱性。

圖1 HPMA制備過程的熱重和微分熱重曲線Fig.1 TG and DTG spectrum of HPMA preparation process
2.4 HPMA制備過程的紅外光譜分析


圖2 HPMA制備過程的紅外光譜Fig.2 FT-IR spectrum of HPMA preparation process
由圖2與表3可知,A、B、C曲線無新峰產生,說明改性只使蛋白質結構發生變化,沒有發生化學反應生成新物質。D在1 740 cm-1處出現1個微弱的吸收峰,可能是PAE中的羥基與花生蛋白中的羧基發生反應,生成酯鍵。D在1 396和1 249 cm-1處的吸收峰面積明顯減少,說明此吸收峰代表的羧酸和氨基與PAE中的氮雜環丁烷發生開環反應,生成交聯網絡結構,進一步佐證D膠合強度提高的原因。
2.5 HPMA制備過程的SEM分析
HPMA制備過程中固化橫截面的微觀形態如圖3所示。A的表面粗糙無序,且褶皺和孔洞較多,水分可以輕易滲透降低耐水性和膠合強度,與未改性大豆蛋白基膠黏劑固化后的斷面相似(Qiangetal.,2012)。與A相比,改性后B的表面更加有序均一,與蛋白質二級結構分析一致,進一步證實 SDS展開了蛋白質二級結構,分子重新有序排列。加入nSiO2后,C的表面凹陷孔洞減小。加入PAE后,D的橫截面更加緊致,粗糙面減少,證實形成超支化網絡結構,有效提高了HPMA的強度和耐水性(Yuanetal.,2017)。

圖3 HPMA制備過程的SEMFig.3 SEM images of HPMA preparation process
由上述熱穩定性、紅外光譜和SEM綜合分析可得HPMA的膠接機理如圖4所示。
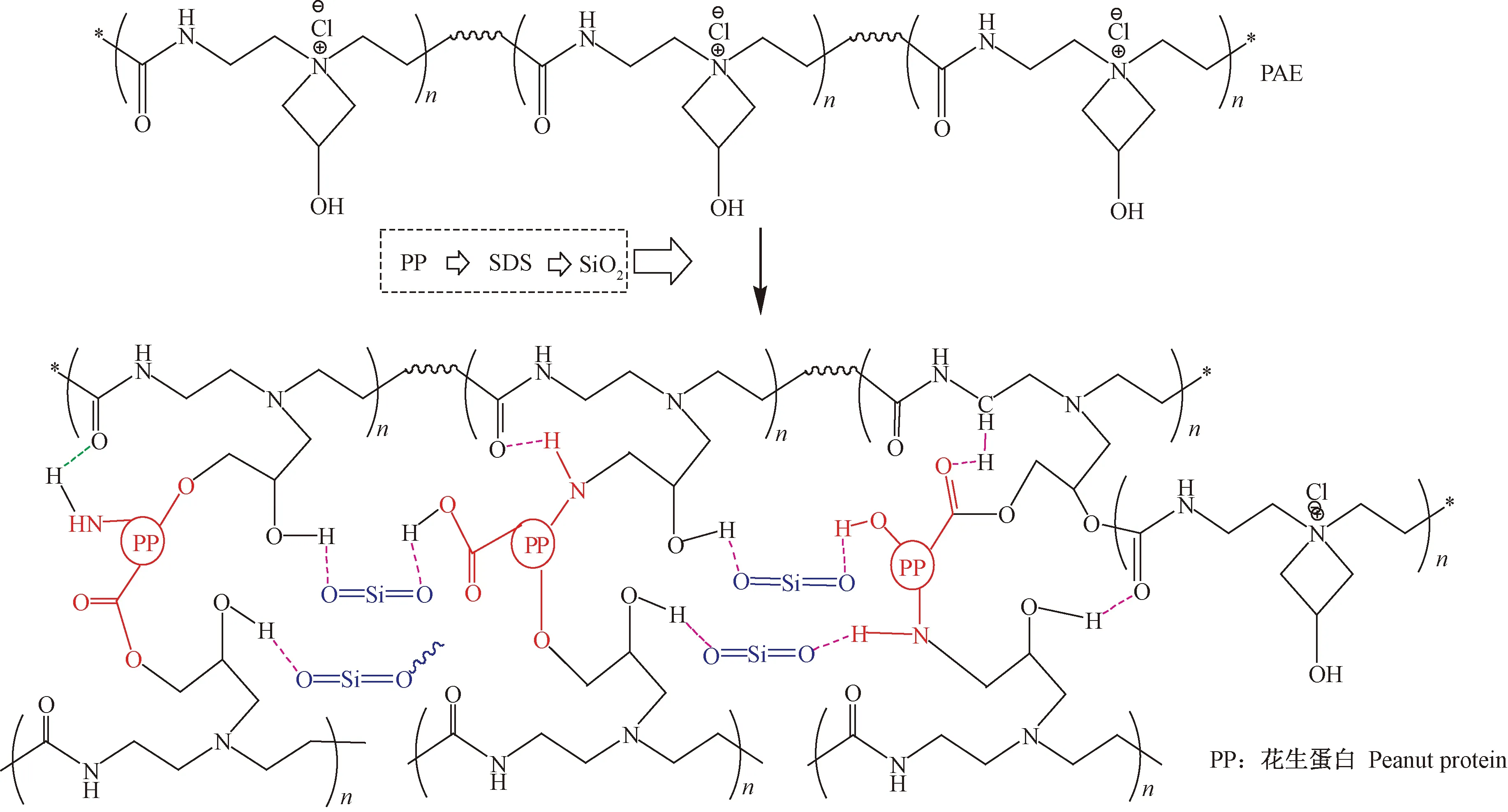
圖4 HPMA制備過程的膠接機理Fig.4 Bonding mechanism of HPMA preparation process
3 結論
本研究以高溫花生粕為試驗材料,采用SDS、nSiO2和PAE三步復合改性交聯制備HPMA,以其制備的楊木膠合板干態、溫水濕態和沸水濕態膠合強度分別提高113%、114%和81%,且沸水濕態膠合強度滿足國家Ⅰ類膠合板標準。HPMA與膠黏劑復合改性交聯前相比,物化性能和耐熱性較好,膠合強度和耐水性提高的主要原因是SDS使蛋白質二、三級結構打開,疏水基團暴露,并與nSiO2和PAE發生超支化交聯形成不溶水的三維網絡結構,本研究結果可為HPMA產業化應用提供技術和理論依據。
猜你喜歡
紡織科學研究(2020年1期)2020-05-21 00:31:06
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2016年2期)2016-06-15 20:30:00
中國塑料(2016年2期)2016-06-15 20:29:59
中國塑料(2016年5期)2016-04-16 05:25:36
廣西林業科學(2016年3期)2016-03-16 05:43:30
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
中國塑料(2015年4期)2015-10-14 01:09:19
