長鏈非編碼RNA 調控神經病理性疼痛的研究進展*
2021-07-30 06:59:00張如月武彩花劉永敏
中國疼痛醫學雜志 2021年7期
張如月 武彩花 李 熳 賈 珉 劉永敏 △
(1 華中科技大學同濟醫學院基礎醫學院,武漢 430030;2 華中科技大學同濟醫學院附屬協和醫院,武漢 430022;武漢市第一醫院3 針灸科;4 檢驗科,武漢 430022)
神經病理性疼痛 (neuropathic pain, NP) 是由軀體感覺神經系統的損傷或疾病直接或間接造成的一種疼痛,其主要表現為自發性疼痛、痛覺過敏、異常疼痛和感覺異常等臨床特征[1]。中樞敏化和外周敏化共同參與神經病理性疼痛的發生發展,其中神經元的異常放電與神經膠質細胞的功能失調改變了傷害性信號的傳導和處理,最終導致痛閾的降低[2]。目前的治療藥物僅能緩解病人的疼痛癥狀,不能改善外周神經的損傷,未能從神經病理性疼痛的發生機制上解決疼痛問題。因此,探究神經病理性疼痛發生發展的分子機制,不僅有利于加深對疼痛本質的認識,對疼痛病人的臨床診治也有重要意義。
長鏈非編碼RNA (long non-coding RNA, lncRNA)是一組長度大于200 個核苷酸、沒有明顯蛋白編碼功能的RNA,由RNA 聚合酶II 催化轉錄合成。越來越多的研究結果表明lncRNA 參與神經元活動以及神經損傷,其中機制可能是通過調節神經元的生長、分化以及突觸的形成和功能來影響神經系統發育和突觸可塑性[3]。已有文獻報道神經病理性疼痛模型中存在著大量lncRNA 的差異表達,干預這些 lncRNA 能有效緩解神經病理性疼痛,由此說明lncRNA 在神經病理性疼痛的發生發展過程中發揮著重要作用。本文就近年來lncRNA 參與神經病理性疼痛的細胞和分子機制及其介導的下游潛在信號通路進行綜述,為深入了解神經病理性疼痛的發生發展提供理論支持,為神經病理性疼痛的臨床治療提供新靶標。
一、lncRNA 與神經病理性疼痛
神經病理性疼痛起源于外周或中樞神經系統損傷(三叉神經痛、脊髓損傷)、病毒感染(帶狀皰疹后神經痛)、代謝紊亂(糖尿病神經病理性疼痛)、卡壓、腫瘤等[4],嚴重影響病人的生活質量。神經病理性疼痛的模型分為外周神經損傷模型和糖尿病神經病理性疼痛模型 (diabetic neurophathic pain,DNP)兩大類。常見的外周神經損傷模型有坐骨神經慢性壓迫損傷模型 (chronic constriction injury, CCI)、坐骨神經切斷模型 (sciatic nerve transaction, SNT)、脊髓神經結扎模型 (spinal nerve ligation, SNL)等。DNP 模型是通過腹腔注射鏈脲佐菌素誘導速發型實驗性糖尿病模型,糖尿病引起的外周神經損傷導致神經病理性疼痛。下面將從目前lncRNA 在調控神經病理性疼痛中的幾個視角分別進行綜述。
1. lncRNA 調控離子通道、興奮神經元促進神經病理性疼痛的發生發展
在神經損傷時,電壓依賴性鉀通道 (Kv) 亞基kcna2 的表達下調,導致DRG 神經元靜息期鉀離子外流減少,靜息電位升高,神經元興奮性升高。Zhao 等[5]發現lncRNA Kcna2-AS (Kcna2 antisense RNA)可以在DRG 神經元沉默Kcna2 進而導致神經病理性疼痛。Kcna2-AS 是 Kcna2 的反義鏈,它的大部分序列與Kcna2 RNA 互補。外周神經損傷時,DRG中 Kcna2-AS 的表達顯著升高,抑制Kcna2 mRNA和蛋白質的表達,使總Kv 電流減少,DRG 神經元的興奮性增加促進神經病理性疼痛的發生。進一步研究發現轉錄因子髓系鋅指蛋白1 (transcriptional activator myeloid zinc finger protein 1, MZF1) 可以與Kcna2-AS 的啟動子區結合,促進 Kcna2 的反義鏈Kcna2-AS 的轉錄,而下調Kcna2-AS 的表達可以緩解神經病理性疼痛。
最近浙江大學Zhang 等[6]的發現脊髓背角lncRNA uc.153 在小鼠CCI 模型中顯著升高,敲低uc.153 的表達可以抑制CCI 導致的脊髓廣動力神經元自發活性和自發放電率的增加,逆轉脊髓敏化,進而改善慢性神經病理性疼痛,并且uc.153 在naive小鼠過表達也會產生痛超敏及脊髓神經元敏化。進一步研究發現pre-miR-182-5p 是其下游靶標,uc.153 負性調控miR-182-5p 及EphB1-NMDA 受體參與慢性神經病理性疼痛的調節。
南 昌 大 學Li等[7]在大鼠CCI模型中發現lncRNA MRAK009713 的表達上調,而抑制MRAK009713的表達可以顯著減輕大鼠痛行為,進一步研究發現MRAK009713 通過與 P2X3 受體相互作用,引起ATP 介導的細胞電流增加,提高了DRG 傷害性神經元的興奮性,增加CCI 大鼠的疼痛敏感行為。在2017 年該 課 題 組 又 發 現 在 2 型 糖 尿 病 大 鼠(type 2 diabetes mellitus, T2DM) 模 型 中 沉 默 lncRNA NONRATT021972[8]可以下調 P2X7 mRNA 及蛋白水平的表達,抑制膠質細胞中BzATP 激活電流,進而抑制DRG 星形膠質細胞的活化以及神經元興奮性,減少炎癥介質的釋放,最終達到緩解T2-DM 神經病理性疼痛的目的。
在臂叢神經撕脫模型 (brachial plexus avulsion,BPA) 中脊髓背角神經元胞漿產生的lncRNA Malat1[9]減少,而在脊髓背角原代培養的神經元中Malat1 的下調導致神經元自發性放電頻率的顯著增加。進一步分析表明在谷氨酸刺激過程中,敲低Malat1 表達的神經元中胞內鈣濃度明顯高于正常神經元。說明Malat1 表達下調可能通過調節鈣電流來增加脊髓神經元興奮性以誘導神經病理性疼痛。
以上研究結果表明,一些lncRNA 與離子通道蛋白或離子通道受體密切相關,外周神經損傷引起這些lncRNA 表達異常,進而使與其相關的離子通道蛋白表達或功能異常,引起神經元或膠質細胞膜電流改變進而影響神經元興奮性,最終導致神經病理性疼痛的發生。
2. lncRNA 通過調節炎癥通路、氧化應激以及自噬調控神經病理性疼痛
外周神經損傷會引起炎癥反應,研究發現lncRNA 可以激活炎癥通路促進炎癥介質釋放促進神經病理性疼痛的發生發展。Yu 等[10]檢測了152 例臨床2 型 糖 尿 病 病 人 的 血 清 ,發 現LncRNA NONRATT021972 和前炎癥因子TNF-α 在 T2DM 組顯著高于對照組,并與神經病理性疼痛的嚴重程度呈正相關,動物實驗也證實沉默NONRATT021972可以降低TNF-α 的表達,減輕炎癥反應,進而緩解神經病理性疼痛。
南昌大學Liang 等[11]在T2DM 大鼠模型中對該LncRNA 研究也得到相同的結論,進一步研究發現NONRATT021972 可能作用于P2X3,促進炎癥介質TNF-α 的釋放,并磷酸化激活ERK1/2 信號通路,促進神經病理性疼痛的發生發展。該課題組還發現沉默lncRNA uc.48+[12]可以抑制DRG 中P2X3受體的表達,使ERK1/2 通路介導的磷酸化和興奮性降低,TNF-α 釋放減少,從而減輕神經病理性疼痛。細胞水平的實驗[13]也發現高糖高游離脂肪酸處理巨噬細胞系RAW264.7 后,uc.48+和P2X7 受體表達升高,P2X7 介導的炎癥介質IL-10 和IL-1β釋放,活性氧 (reactive oxygen species, ROS) 形成,ERK 通路被激活,而沉默uc.48+可以翻轉P2X7 的表達并抑制其介導的炎癥和免疫反應。Liu 等[14]研究發現T2DM 大鼠模型中lncRNA BC168687 高表達,瞬時受體電位香草酸亞型1 (transient receptor potential channel subfamily vanilloid 1, TRPV1) 受體被激活,磷酸化的ERK 通路和磷酸化的p38 信號通路同時被激活,血清炎癥因子TNF-α 和 IL-1 升高,促進神經病理性疼痛的發生發展,而下調 lncRNA BC168687 可以減輕TRPV1 介導的糖尿病神經病理性疼痛。該課題組在細胞水平的研究結果也發現高糖高游離脂肪酸處理星形膠質細胞后,lncRNA BC168687 和 P2X7 升 高,siRNA 下 調 BC168687 可降低P2X7 的表達以及P2X7 介導的NO 和 ROS 的形成,減輕氧化應激[15]。
最新研究發現lncRNA 可以通過調節細胞自噬參與神經病理性疼痛。CircRNAs (CircularRNAs, 環狀RNA) 是一類不具有5'末端帽子和3'末端poly (A)尾巴、并以共價鍵形成環形結構的客觀存在于生物體內的非編碼RNA 分子,它也屬于lncRNA。有研究表明,環狀RNA 也參與神經病理性疼痛的調節,Cai等[16]發現ciRS-7 (circRNA-7) 在神經病理性疼痛中表達上調,ciRS-7 的表達與CCI 神經病理性疼痛的進展正相關。ciRS-7 可以部分上調CCI 大鼠自噬和炎癥表達水平,而下調ciRS-7 的表達可以減輕自噬和炎癥進而改善神經病理性疼痛,進一步研究發現ciRS-7 是通過海綿吸附miR-135a-5p 調節神經病理性疼痛。
大量文獻表明神經病理性疼痛的發生發展與免疫反應、炎癥反應的激活密切相關。神經病理性疼痛時lncRNA 可以通過調節神經炎癥、氧化應激、自噬等生物學過程,影響炎癥介質、ROS 以及自噬生物標志物合成或釋放,使神經病理性疼痛得以維持和發展。
3. lncRNA 調節神經遞質釋放參與神經病理性疼痛
吉林大學Li 等[17]發現脊髓背角lncRNA H19在神經病理性疼痛模型高表達,神經遞質5-羥色胺(5-hydroxytryptamine, 5-HT)、GABAB2在脊髓背角低表達。進一步研究發現,H19 通過海綿吸附miR-196a-5P 調控細胞周期蛋白依賴性激酶5 (cyclin-dependent kinase 5, CDK5)/P35 到磷酸化的cAMP 反應元件結合蛋白 (p-CREB) 信號通路(CDK5/P35/CaMKII/p-CREB),最終影響神經遞質5-TH、GABAB2和炎癥介質的合成。抑制性遞質合成減少、炎癥介質釋放增多共同促進神經病理性疼痛的發生發展。
4. lncRNA 調節神經元凋亡參與神經病理性疼痛
昆明醫科大學Liu 等[18]研究發現在臂叢神經損傷 (acute brachial plexus injury, BPI) 模型中,lncRNA JHDM1D-AS 1和DUSP1(又被稱為MAPK磷酸酶-1,MKP-1)表達顯著下降,miR-101-3p 表達升高。過表達JHDM1D-AS 1 抑制BPI 引起的神經元凋亡和小膠質細胞的激活。進一步研究發現,在BPI 模型中,調控JHDM1D-AS1/miR-101-3p/DUSP1/P38/NF-KB 信號通路可引起脊髓背角小膠質細胞的過度激活(神經炎癥)和神經元凋亡。而JHDM1D-AS1 正是通過吸附miR-101-3p 進而上調DUSP1 來抑制P38/NF-κB途徑的激活來發揮神經保護以及抗炎作用。
5.神經病理性疼痛中lncRNA、miRNA 以及mRNA 的相互作用
近幾年報道了大量長鏈非編碼RNA 通過lncRNA-miRNA-mRNA 軸調節神經病理性疼痛發生發展的研究,經過梳理,lncRNA, miRNA 以及mRNA 的相互作用機制如下:①lncRNA 作為分子海綿,吸附miRNA,抑制其與mRNA 的結合,使得mRNA 免于降解,這類lncRNA 被稱為內源競爭RNA (competing endogenous RNAs, ceRNA)[19~22];②miRNA 可引起lncRNA 降解。miRNA 通過減弱靶lncRNA 的穩定性來降低其豐度,進而影響多種生物學過程。例如,miR-145-5p 是人類胚胎干細胞中lncRNA-RoR 的靶基因,而增加miR-145-5p 的濃度會降低lncRNA-RoR 的活性[23];③lncRNA 與miRNA 競爭性結合mRNA,lncRNA 也可以直接在miRNA-mRNA 結合位點區域與mRNA 結合,從而消除miRNA 對mRNA 的調控[24];④lncRNA 可以產生miRNA,lncRNA 可能含有發夾結構,可以產生pre-miRNA(miRNA 前體),進一步調節下游基因的表達[25]。在神經病理性疼痛的研究中,目前已報道的多為lncRNA 通過海綿作用吸收miRNA進而中和miRNA 對靶基因的作用。多項研究證明lncRNA XIST 在CCI 動物模型中表達明顯上調,通過調控下游靶miRNA miR-150[22],miR-137[19],miR-154-5p[20],miR-544[21]的表達,進而作用于核轉錄因子STAT3、ZEB1 以及TLR5 影響細胞的炎癥信號通路、氧化應激等促進神經病理性疼痛的發生發展。近幾年報道了很多 lncRNA 通過調控microRNA 的表達進而影響炎癥介質釋放、氧化應激來參與神經病理性疼痛的研究,具體機制見表1[26~37]。
長鏈非編碼RNA 在神經病理性疼痛模型中異常表達,通過一系列分子機制影響神經元或膠質細胞的功能,調控神經病理性疼痛的發生發展。表1從模型、下游分子機制,功能等方面匯總了近年來報道的lncRNA 參與神經病理性疼痛的研究。
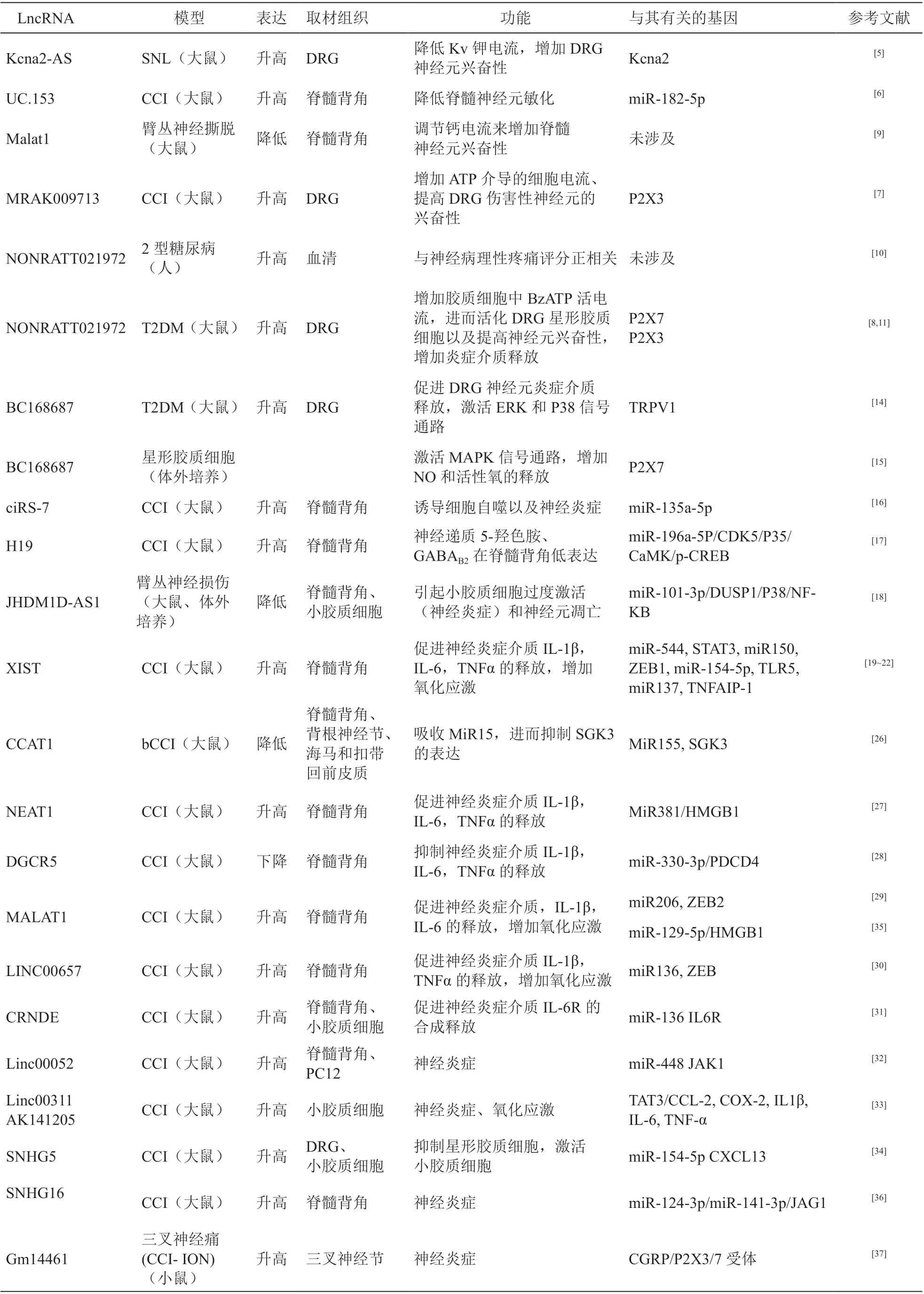
表1 lncRNA 在神經病理性疼痛中的功能特征匯總
二、神經病理性疼痛模型中差異lncRNA 的表達譜分析
隨著RNA 測序技術的發展和普及,神經病理性疼痛模型的差異lncRNA 及mRNA 被篩選出來并得到驗證。下面匯總了近幾年發表的神經病理性疼痛模型RNA 測序、差異基因表達譜分析的研究(見表2)。
1. 脊神經結扎模型,研究的側重點在神經病理性疼痛相關的lncRNAs
Jiang 等[38]對大鼠實施脊神經結扎術 (spinal nerve ligation, SNL) 后 10 天取 lncRNA 和 mRNA 基因芯片分析,發現511 個差異表達的lncRNA(其中 366 個表達上調、145 個表達下調)和 493 個差異表達的mRNA(其中 363 個表達上調、122 個表達下調)。對上調的 lncRNA 和 mRNA 進行功能分析,發現差異表達的 mRNA 功能主要集中在免疫反應、防御反應和炎癥反應,這也是神經病理性疼痛
的重要致病機制。進一步分析發現有35 個差異表達的lncRNA 與差異表達的 mRNA 相鄰或重疊,功能分析發現其與toll 樣信號受體、細胞因子受體相互作用、過氧化物酶體增殖物激活的受體信號通路有關(見表2)。
新澤西州醫學部Wu 等[39]對小鼠脊神經結扎模型6 天后 L4DRG 進行RNA 測序,發現了944 個差異表達的的非編碼RNA(見表2),其中變化最顯著的是長鏈固有非編碼RNA,其次是反義RNA、假基因。后期qPCR 驗證 Kcna2, Oprm1 以及lncRNAs Gm21781 和4732491K20Rik 在脊神經結扎后確實存在差異表達。
2. 坐骨神經損傷模型,研究的側重點在與神經再生相關的lncRNAs
Yu 等[40]對大鼠坐骨神經切斷后0、1、4、7 天的背根神經節 (dorsal root ganglion, DRG) 進行RNA芯片分析,發現了105 個差異表達的lncRNA,其中有24 個下調的lncRNAs 以及115 個靶基因,通過信號通路分析發現下調的lncRNA 與神經膠質細胞遷移、嘌呤能核苷酸受體信號通路、血管舒張、神經肽信號通路以及神經再生信號通路包括 MAPK信號通路和神經活性配體-受體相互作用有關(見表2),其中驗證了沉默lncRNA BC089918 可以促進外周神經損傷后軸突再生。而Yao 等[41]也通過RNA 表達譜分析發現lncRNAs uc.217 在外周神經再生中有重要調控作用,沉默uc.217 促進神經元的軸突再生,通過生物信息學分析和實驗驗證確定uc.217 靶向蛋白質sema3d 和smad7 參與了神經再生。Mao 等[42]對大鼠坐骨神經損傷模型7 天后L4-L6DRG 轉錄組水平進行高通量測序,發現了86 個已知的 lncRNA 和 26 個新的lncRNA 存在差異表達,對這86 個已知的lncRNA 所調控的866 個靶基因進行GO 分析和KEGG 富集分析發現單側坐骨神經損傷后涉及到神經元合成、分泌神經遞質的基因表達下調,參與神經元再生的基因表達上調,說明神經再生成為應對外周神經損傷后的中堅力量。RTqPCR 驗證了下調的rno-Cntnap lncRNA 和上調的AC111653.1 lncRNA,說明它們與神經病理性疼痛有關,并推斷它們可以促進周圍神經再生。
3. 糖尿病神經病理性疼痛模型中差異lncRNA的表達譜分析
糖尿病周圍神經病變是糖尿病病人最常見、最復雜和最嚴重的并發癥之一,是因糖尿病慢性高血糖狀態及其所致各種病理生理改變而導致的神經系統損傷,臨床上多表現為肢體疼痛、感覺減退、麻木、灼熱、冰涼等,也可表現為自發性疼痛、痛覺過敏、痛覺超敏。與其他類型的神經性疼痛病人相似,病人表現出外周和中樞敏化,病理性小膠質細胞活化和參與傷害性感受的神經元和神經膠質細胞的突觸可塑性變化。Du 等[43]通過對鏈脲佐菌素 (streptozotocin, STZ) 誘導的DNP 小鼠脊髓背角組織RNA芯片分析發現有1481 個差異表達的 lncRNAs 和 1096個差異表達的mRNA,功能分析發現TGF-β 結合是最重要的分子功能,逆行的內源性大麻素信號轉導通路是差異mRNA 最重要的通路,其次是鈣離子轉運(見表2)。最后驗證ENSMUST00000150952-Mbp和AK081017-Usp15 與DNP 的發生發展密切相關。

表2 神經病理性疼痛模型中lncRNAs 的差異表達譜匯總
三、結語
通過RNA 測序和RT-qPCR,越來越多參與神經病理性疼痛的lncRNA 被篩選出來并得到驗證,并且它們的功能和調控機制也在被探索。從功能機制上看,lncRNA 主要是通過離子通道影響神經元興奮性以及神經炎癥氧化應激這兩方面參與神經病理性疼痛,而最新研究發現lncRNA 也可以通過調節神經元凋亡、神經遞質合成釋放等其他角度參與神經病理性疼痛。從分子機制上看,lncRNA 主要是通過其反義RNA(如KCNA2-AS 和JHDM1DAS1)與mRNA 結合引起mRNA 降解以及lncRNA海綿吸附miRNA 進而調節下游靶基因參與神經病理性疼痛。這些研究為它們在神經病理性疼痛的臨床診斷和治療中作為潛在的治療靶標提供了新的啟示。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
光學精密工程(2016年6期)2016-11-07 09:07:19
