人工DNA合成技術(shù):DNA數(shù)據(jù)存儲的基石
2021-07-21 09:30:44黃小羅戴俊彪
合成生物學(xué) 2021年3期
關(guān)鍵詞:方法
黃小羅,戴俊彪
(中國科學(xué)院深圳先進技術(shù)研究院,深圳合成生物學(xué)創(chuàng)新研究院,廣東省合成基因組學(xué)重點實驗室,深圳市合成基因組學(xué)重點實驗室,廣東 深圳 518055)
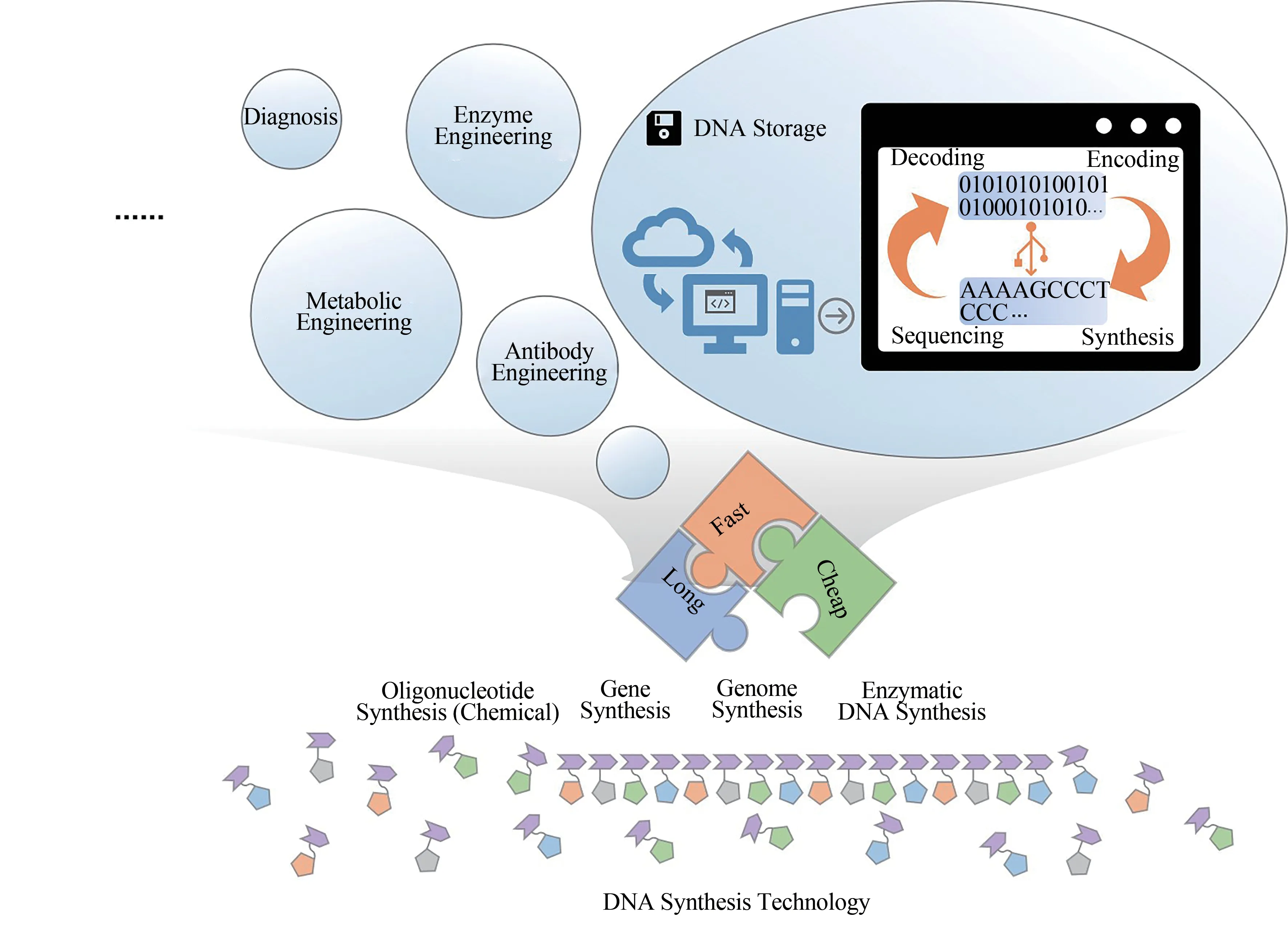
互聯(lián)網(wǎng)、人工智能、大數(shù)據(jù)領(lǐng)域的發(fā)展催生了全球數(shù)據(jù)的指數(shù)級增長。以目前的發(fā)展趨勢,傳統(tǒng)的數(shù)據(jù)存儲介質(zhì),比如硬盤、磁帶、光盤,將無法應(yīng)對不斷增長的數(shù)據(jù)需求。DNA作為一種存儲介質(zhì),具有存儲密度高、存儲時間長、占地面積小、維護成本低等諸多優(yōu)點,是解決未來數(shù)據(jù)存儲危機最有潛力的介質(zhì)之一[1-2]。自2012年Church等[3]發(fā)表基于DNA介質(zhì)的下一代數(shù)據(jù)存儲研究開始,越來越多的研究工作開始聚焦在這一領(lǐng)域。目前已發(fā)表的DNA數(shù)據(jù)存儲工作中[3-6],其流程大致包括如下幾個主要的步驟:①數(shù)據(jù)編碼,從存儲在計算機上的數(shù)據(jù)中提取二進制0/1信息,并通過設(shè)計的0/1二進制信息到A/T/C/G序列的映射關(guān)系,將0/1二進制信息轉(zhuǎn)換為A/T/C/G堿基序列;②DNA合成,利用人工DNA合成技術(shù)將堿基序列合成為可以被保存的DNA多聚物分子;③DNA測序,利用測序技術(shù)讀取合成的DNA多聚分子的堿基序列;④數(shù)據(jù)恢復(fù),利用步驟①中設(shè)計的0/1二進制信息到A/T/C/G序列的映射關(guān)系,將步驟③中獲得的DNA序列轉(zhuǎn)換為0/1二進制信息,并進一步轉(zhuǎn)換成為存儲的數(shù)據(jù)。人工DNA合成技術(shù)作為DNA存儲流程中的核心技術(shù),是DNA數(shù)據(jù)存儲從概念走向應(yīng)用的關(guān)鍵。
人工DNA合成技術(shù),是在不依賴DNA模板的情況下,根據(jù)人為設(shè)計的任意序列進行DNA合成的技術(shù)。1953年,沃森和克里克[7]發(fā)現(xiàn)了DNA雙螺旋結(jié)構(gòu),使得人們對DNA分子的認知實現(xiàn)了一個里程碑式的跨越。自此開始,一批化學(xué)家和生物學(xué)家們就開始了人工DNA合成的研究。目前的人工DNA合成技術(shù),不僅能夠合成幾十到數(shù)百個堿基的寡聚核苷酸,而且能夠通過化學(xué)合成、結(jié)合酶法拼接及微生物克隆等方法,合成Mb級別的微生物基因組。DNA合成也從實驗室走向了商業(yè)化。GE Healthcare(美國)、Biolytic(美國)、BioAutomation(美國)等公司推出一系列不同通量的合成儀;提供DNA合成服務(wù)的公司也逐漸發(fā)展起來,包括IDT(美國)、GenScript(美國/中國)、GeneWiz(美國)、Twist Bioscience(美國)等。這些公司也在DNA合成技術(shù)開發(fā)、成本降低以及支撐DNA合成下游應(yīng)用方面做出了重要貢獻。
除DNA數(shù)據(jù)存儲外,人工合成DNA在醫(yī)藥、農(nóng)業(yè)、材料等多個合成生物學(xué)支撐的領(lǐng)域都發(fā)揮著重要的作用。在新冠肺炎[8]、乙肝[9]、埃博拉病毒病[10]等多種公共傳染疾病的防控上,人工合成DNA探針被用作核酸檢測試劑盒的關(guān)鍵原料。在醫(yī)藥開發(fā)上,人工合成DNA極大加速了生物抗體藥改造[11]、基因治療[12-13]、寡核苷酸藥物[14]等開發(fā)過程,是這些藥物應(yīng)用不可或缺的技術(shù)手段。在農(nóng)業(yè)育種上,人工合成DNA可以用于轉(zhuǎn)基因農(nóng)作物品種的改造,如將人工改造合成的來源于蘇云金芽孢桿菌殺蟲蛋白Cry的編碼基因轉(zhuǎn)化到農(nóng)作物體內(nèi)[15],產(chǎn)生了抗蟲棉、抗蟲稻等系列抗蟲品種。在材料領(lǐng)域,基于人工合成DNA制備的DNA納米機器人被報道成功地將凝血酶帶到腫瘤細胞,殺死腫瘤細胞[16]。DNA人工合成技術(shù)對合成生物學(xué)領(lǐng)域的支撐作用,堪比測序技術(shù)對基因組學(xué)和精準醫(yī)學(xué)領(lǐng)域的貢獻,是合成生物學(xué)發(fā)展的關(guān)鍵技術(shù)。
本文總結(jié)了目前人工DNA合成的關(guān)鍵技術(shù)研究進展,包括寡核苷酸合成、基因合成、基因組合成以及新一代酶法DNA合成等。與此同時,本文進一步討論了人工DNA合成技術(shù)在DNA數(shù)據(jù)存儲中的應(yīng)用。
1 寡核苷酸化學(xué)合成技術(shù)
在人工合成DNA中,單鏈寡核苷酸的應(yīng)用形式最為廣泛,比如PCR引物、NGS(next generation sequencing)捕獲探針文庫、寡核苷酸藥物等。自從20世紀50年代,Todd等[17]合成出第1個二嘧啶核苷[d(TpT)和d(pTpT)],一系列寡核苷酸合成方法被開發(fā)出來,包括磷酸二酯法、磷酸三酯法、亞磷酸三酯法、亞磷酰胺法等[18-23]。其中,20世紀80年代發(fā)展起來的固相亞磷酰胺化學(xué)法[18-19,23]被廣泛應(yīng)用于各商業(yè)化的自動化合成儀開發(fā)中。目前大多使用的亞磷酰胺化學(xué)寡核苷酸合成法包括如下4個步驟(圖1):
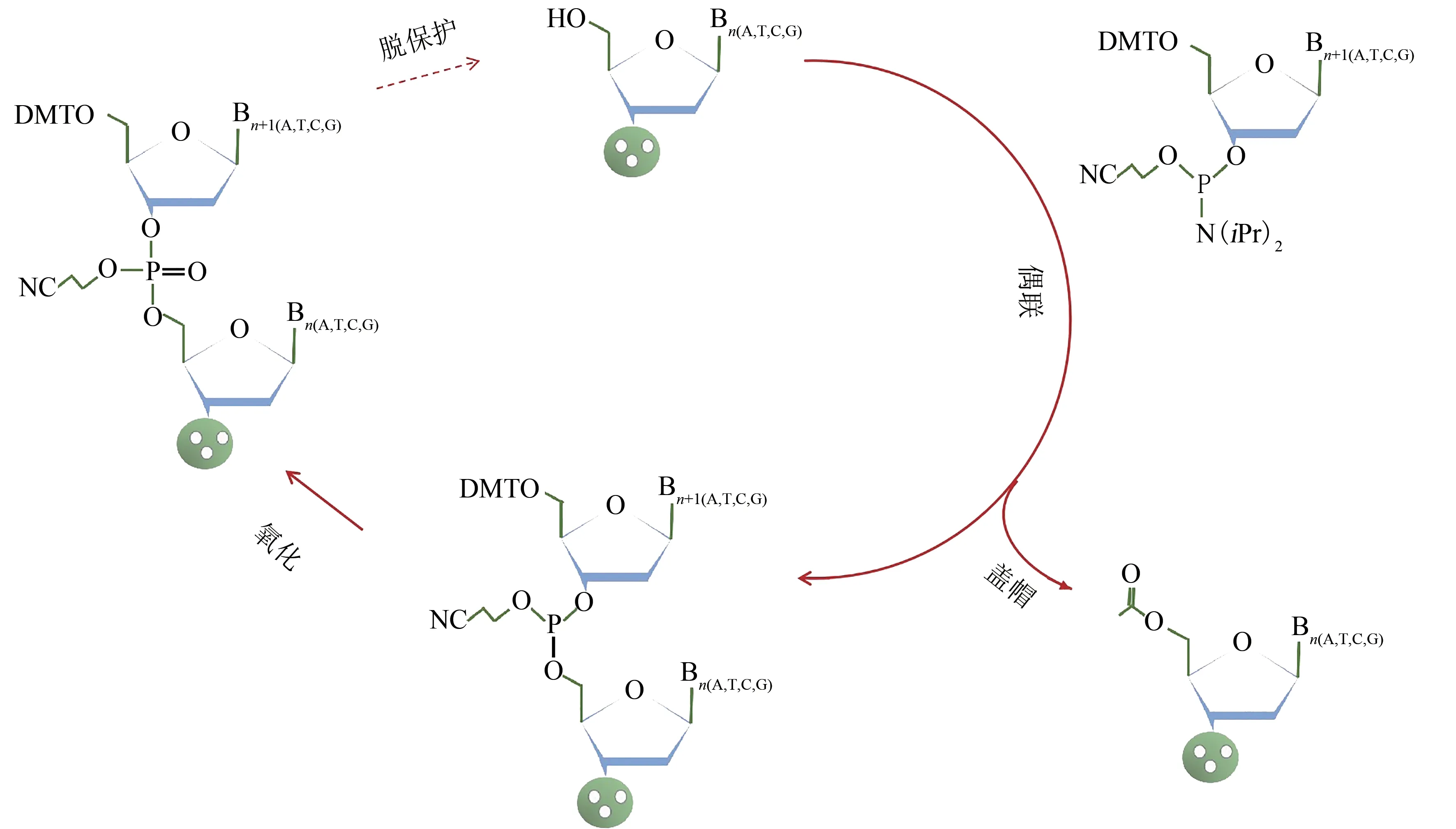
圖1 亞磷酰胺四步化學(xué)寡核苷酸合成法[18-19,23,90]Fig.1 Oligonucleotides synthesis based on four-step"phosphoramide"method[18-19,23,90]
(1)脫保護:將連接在固相載體上的亞磷酰胺核苷上的保護基團DMT(dimethoxytrityl)基團,通過三氯乙酸的處理去除掉,獲得游離的5′-羥基。
(2)偶聯(lián):新的DMT保護的亞磷酰胺核苷通過與四氮唑混合進行活化,得到活化的3′端,與上一個亞磷酰胺核苷的游離5′-羥基發(fā)生縮合反應(yīng)。
(3)蓋帽:步驟2中沒有偶聯(lián)成功的5′-羥基,通過加入乙酸酐和N-甲基咪唑進行乙酰化反應(yīng),避免與后續(xù)堿基的偶聯(lián)反應(yīng),減少寡核苷酸合成過程中的刪除錯誤。
(4)氧化:通過氧化劑碘的作用,將亞磷形式轉(zhuǎn)化為穩(wěn)定的五價磷形式。
通過以上4個步驟的循環(huán),將與預(yù)定合成寡核苷酸序列一致的堿基,通過3′→5′的方式一個個延伸合成。根據(jù)實現(xiàn)方式的不同,寡核苷酸化學(xué)合成技術(shù)主要包括柱式寡核苷酸合成以及芯片寡核苷酸合成。
1.1 柱式寡核苷酸化學(xué)合成
柱式寡核苷酸化學(xué)合成利用一個帶有反應(yīng)腔的合成柱,裝載用于寡核苷酸合成的固相載體,配合流體系統(tǒng),來實現(xiàn)化學(xué)寡核苷酸合成的四步循環(huán)反應(yīng)。目前常用固相載體為可修飾的多孔玻璃(controlled pore glass,CPG)載體[24-25],通過高分子聚乙烯等材料的顆粒包埋而成。而固相載體材料內(nèi)部的孔腔,能夠允許亞磷酰胺化學(xué)寡核苷酸合成的四步化學(xué)反應(yīng)的試劑在其中流動,并依賴于CPG載體,將修飾的亞磷酰胺堿基一個個合成上去。寡核苷酸在合成柱上完成合成后,通過能破壞固相載體和寡核苷酸之間連接間臂的化學(xué)反應(yīng)從合成柱上切割下來。根據(jù)合成柱體積以及其中帶有的CPG載體量的不同,合成寡核苷酸的量也表現(xiàn)出很大的差別,從pmol到mmol級別不等。
柱式寡核苷酸化學(xué)合成是目前多款商用自動化合成儀采用的主要方法。目前的柱式寡核苷酸合成儀,比較成熟的有Biolytic公司開發(fā)的Dr.Oligo系列合成儀以及BioAutomation公司開發(fā)的Mermade系列合成儀。這些儀器能夠?qū)崿F(xiàn)48~1536個單合成柱的寡核苷酸并行合成。通過電磁閥,能夠精確控制反應(yīng)試劑流入合成柱,單個合成柱的產(chǎn)物規(guī)模通常在0.5~10 nmol。實驗室經(jīng)常使用的PCR引物、qPCR探針等,就是用這類合成儀進行合成。另外一種柱式合成儀單次合成寡核苷酸量相對較大,比如GE公司的Oligo-Plot系列,單柱的合成量多達30 mmol,主要用于寡核苷酸藥物等大規(guī)模寡核苷酸原料的制備。
柱式寡核苷酸合成技術(shù)目前相對成熟,其應(yīng)用也較為廣泛。經(jīng)過幾十年的發(fā)展,通過儀器、軟件、反應(yīng)程序、反應(yīng)步驟、純化步驟等的優(yōu)化,目前,脫鹽純化的單鏈PCR用寡核苷酸,單堿基市售價格能夠低至0.3~0.6元/堿基。然而,柱式合成的寡核苷酸由于每輪合成反應(yīng)效率通常低于99.5%,柱式合成120 nt以上、高純度的引物非常困難,這一定程度上限制了柱式合成寡核苷酸的應(yīng)用。因此,開發(fā)更長的單鏈柱式寡核苷酸合成技術(shù)對于進一步提升柱式寡核苷酸的應(yīng)用將具有重要價值。
1.2 芯片寡核苷酸化學(xué)合成
不同于柱式合成,芯片合成中寡核苷酸的化學(xué)合成反應(yīng)在修飾芯片載體上完成。從20世紀90年代Affymetrix的科學(xué)家開發(fā)出寡核苷酸芯片合成技術(shù)開始[26-27],芯片合成技術(shù)通過幾十年的發(fā)展逐漸走向成熟。為了實現(xiàn)高通量并行的寡核苷酸化學(xué)合成,芯片合成技術(shù)需要保證在一個非常小的芯片位點上,能夠不受干擾地單獨完成每一輪的化學(xué)反應(yīng)。為了實現(xiàn)這一目的,高通量光脫保護芯片合成技術(shù)[28-29]、電化學(xué)脫保護芯片合成技術(shù)[30]及噴墨打印合成技術(shù)[31-33]等被開發(fā)出來,這些技術(shù)同時被LC Science(美國)、CustomArray(美國)、Twist Bioscience(美國)進一步拓展及商業(yè)化。從設(shè)計思路上說,這些技術(shù)通過在芯片的點陣上,獨立實現(xiàn)合成脫保護和偶聯(lián)的過程,從而達到在芯片上高通量并行合成的目的;同時,因為芯片合成中單個反應(yīng)體積小,從而極大減少試劑的消耗,實現(xiàn)低成本合成的目的。
高通量芯片寡核苷酸合成能夠一次合成寡核苷酸多達數(shù)十萬條,而成本僅是柱式合成的1/104到1/102。不同于柱式合成,合成出來的寡核苷酸是每條單獨存在,高通量芯片合成的寡核苷酸通常以混合庫的形式存在;同時,合成的混合庫中單條寡核苷酸的量也遠遠低于柱式合成,從fmol到pmol不等。這一定程度上也限制了芯片寡核苷酸合成的應(yīng)用。目前的芯片寡核苷酸合成主要用于突變體庫構(gòu)建、探針捕獲文庫、CRISPR文庫構(gòu)建等對合成量要求不高但序列種類復(fù)雜的領(lǐng)域。
盡管芯片合成寡核苷酸技術(shù)已經(jīng)實現(xiàn)了一定程度上高通量、低成本的合成,然而,相比較高通量DNA測序技術(shù),其通量要低4個數(shù)量級以上,單堿基成本高出5個數(shù)量級以上。這一定程度上制約了其在合成生物學(xué)領(lǐng)域的進一步大規(guī)模應(yīng)用。因此,如何實現(xiàn)高通量寡核苷酸合成通量的進一步提升,將是這一領(lǐng)域技術(shù)發(fā)展面臨的重要難題。
1.3 寡核苷酸的純化方法
由于四步寡核苷酸化學(xué)合成法的每一輪化學(xué)合成的反應(yīng)效率無法保證100%,同時合成過程中也伴隨有一些副反應(yīng)的發(fā)生,合成的產(chǎn)物中摻雜有比目的寡核苷酸序列短的寡核苷酸產(chǎn)物以及其他化學(xué)反應(yīng)副產(chǎn)物。因此合成的寡核苷酸產(chǎn)物,通常還需要進一步純化,以除去化學(xué)反應(yīng)中的短的寡核苷酸和化學(xué)反應(yīng)副產(chǎn)物。
常用的純化方法包括直接脫鹽純化、OPC(oligonucleotide purification cartridge)柱純化、PAGE(polyacrylamide gel electrophoresis)純化、HPLC(high performance liquid chromatography)純化等。
直接脫鹽純化,是將從固相載體上切割下來的引物,通過反復(fù)地溶劑洗脫,去除合成引物化學(xué)反應(yīng)過程中產(chǎn)生的各種鹽類,獲得合成的寡核苷酸混合物。這種純化方式,幾乎無法有效地去除寡核苷酸中短鏈的產(chǎn)物,因此多用于對純度要求不高的寡核苷酸純化。
OPC柱是一種填有對帶有5′-DMT基團寡核苷酸特異吸附能力的純化柱。通過OPC柱對最后一個堿基帶有5′-DMT基團(在合成步驟保留)寡核苷酸的特異吸附,去除截短的寡核苷酸。純化的帶5′-DMT基團的目的寡核苷酸最后通過酸處理除去DMT基團,獲得目的寡核苷酸[34]。這種方法雖然在理論上能夠很好地提高目的寡核苷酸的純度,但是在實際應(yīng)用中,要獲得很好的純化效果,需要充分的優(yōu)化。
PAGE純化是利用變性聚丙烯酰胺凝膠電泳對寡核苷酸進行純化的方法[35]。優(yōu)化的聚丙烯酰胺凝膠電泳能夠很好地分辨不同長度的合成單鏈寡核苷酸。通過電泳后,將目的寡核苷酸條帶切割下來,再通過溶劑將寡核苷酸從膠中釋放出來,能夠獲得純度較高的目的寡核苷酸。這種方法可以用于不同長度、不同應(yīng)用場景的寡核苷酸純化。
HPLC純化是利用C18或者離子交換色譜柱,能夠獲得較其他方法更高的純度[36]。分子生物學(xué)試驗中常用的qPCR探針、NGS探針等多采用這種方法純化。然而,HPLC純化方法多適用于小于100個堿基的寡核苷酸純化。
雖然目前的純化方法能夠?qū)崿F(xiàn)不同應(yīng)用場景的寡核苷酸純化,但是該步驟通常耗費大量的人力,對于超過100個堿基的高純度寡核苷酸純化也比較困難,同時通量較低,因此需要開發(fā)更加高效的寡核苷酸純化方法。另外,對于芯片寡核苷酸合成產(chǎn)物純化,由于合成產(chǎn)物是混合物,而且單條寡核苷酸的量非常少,寡核苷酸產(chǎn)量也不均一,開發(fā)高純度芯片寡核苷酸純化技術(shù)也是高通量芯片寡核苷酸合成需要重點解決的問題。
值得提及的是,化學(xué)寡核苷酸合成常常伴隨著較高的錯誤率。由于蓋帽不充分、反應(yīng)試劑純度不夠、反應(yīng)環(huán)境濕度太高、酸處理時間過長、偶聯(lián)時間不夠等多方面原因[37-39],合成會出現(xiàn)堿基缺失、突變等多種錯誤。實驗室優(yōu)化的柱式化學(xué)合成的錯誤率通常在1/1000到1/500之間;芯片合成的錯誤率則通常在1/500到1/200之間。這一定程度上給化學(xué)合成寡核苷酸的應(yīng)用造成了困擾,比如一些PCR引物克隆試驗,出現(xiàn)了引入插入序列的錯誤問題。因此,未來的寡核苷酸化學(xué)合成技術(shù)可以關(guān)注更高保真度的合成純化方法。
2 基因合成技術(shù)
不同于遺傳學(xué)中“基因”的概念,合成基因中,“基因”主要是指體外合成的雙鏈DNA片段或者克隆到質(zhì)粒載體上的雙鏈DNA,可以包含任意的長度。合成基因在酶工程定向進化、代謝工程改造、抗體工程等多個方向擁有很廣泛的用途。最早的人工體外基因合成研究可以追溯到20世紀60~70年代,利用酶拼接實現(xiàn)從化學(xué)合成寡核苷酸到短的tRNA基因的合成[40-43]。從此之后,以單鏈寡核苷酸為基礎(chǔ)原料,不同長度的基因合成拼接技術(shù)方法被開發(fā)出來。根據(jù)基因合成的技術(shù)步驟先后順序,基因合成可以包括寡核苷酸拼裝、基因合成糾錯與克隆篩選以及大片段基因合成組裝。
2.1 寡核苷酸拼裝技術(shù)
如何從單鏈寡核苷酸拼接出雙鏈DNA是基因合成的第一步。最早使用的寡核苷酸拼接方法,是連接酶依賴的寡核苷酸拼接方法。通過對單鏈寡核苷酸的5′末端進行磷酸化,然后依賴于連接酶,將完全互補配對的寡核苷酸拼接在一起。由于合成錯誤的堿基無法進行互補配對,這種方法能夠?qū)崿F(xiàn)較高保真度的寡核苷酸基因拼接。一系列的研究闡述了利用連接酶依賴的寡核苷酸拼接方法來實現(xiàn)基因組裝[44-46]。最早的寡核苷酸的拼接,是一段段逐漸合成[40-43];然而直到“鳥槍”連接法的發(fā)明,使得多段DNA同時在一個反應(yīng)混合物中組裝成為可能[45-46]。連接酶鏈式反應(yīng)(ligase chain reaction,LCR)技術(shù)的發(fā)明[47]進一步推進了基于連接酶的基因合成技術(shù)的應(yīng)用[48]。根據(jù)連接酶的反應(yīng)特點,片段與片段之間的連接效率是有限的,導(dǎo)致拼接的目的產(chǎn)物寡核苷酸片段產(chǎn)物相對較少。因此,通過連接酶拼接方法和PCR技術(shù)的結(jié)合,利用PCR技術(shù)對連接酶拼接的產(chǎn)物進行擴增,可以獲得大量的寡核苷酸拼接產(chǎn)物[48-49]。
依賴于連接酶的基因合成組裝技術(shù)常常需要合成正反兩條完全互補的寡核苷酸鏈,才能完成基因合成,同時要求寡核苷酸5′端磷酸化,這常常造成合成成本的上升。基于重疊延伸PCR的寡核苷酸組裝技術(shù),聚合酶鏈式組裝技術(shù)(polymerase chain assembly,PCA)能夠一定程度解決這個問題。它通過合成包含堿基重疊區(qū)的基因正負鏈的寡核苷酸,利用優(yōu)化的PCR程序和酶反應(yīng)體系,將寡核苷酸拼接成一條完整的鏈。通過優(yōu)化重疊區(qū)的堿基個數(shù),比如僅使用15~25個堿基,能夠減少一定量化學(xué)寡核苷酸堿基合成。為了提高PCR拼接的成功率,一系列優(yōu)化的方法被發(fā)展出來,比如利用不對稱PCR、系列PCR反應(yīng)擴增等[50-53]。最近十幾年,隨著PCR儀器和PCR試劑價格的降低,PCA技術(shù)被廣泛應(yīng)用于工業(yè)化的基因合成應(yīng)用中。
此外,林繼偉等[54-56]發(fā)明了一種基于等溫延伸的基因合成方法,利用獨特設(shè)計的寡核苷酸,在等溫聚合酶、限制性內(nèi)切酶、外切核酸酶或者連接酶的復(fù)合物的共同作用下,將多條單鏈的發(fā)夾寡核苷酸,組裝延伸成為一條雙鏈的基因。其特殊設(shè)計的寡核苷酸上帶有一個二類限制性內(nèi)切酶的酶切位點,同時5′端帶有一段與3′端互補配對的堿基,可以理論上形成帶有幾個3′端堿基懸掛的特殊發(fā)夾結(jié)構(gòu)(圖2)。這種方法,由于所有的反應(yīng)在等溫條件下完成,操作簡便,可以用于大規(guī)模自動化的寡核苷酸基因合成拼裝。
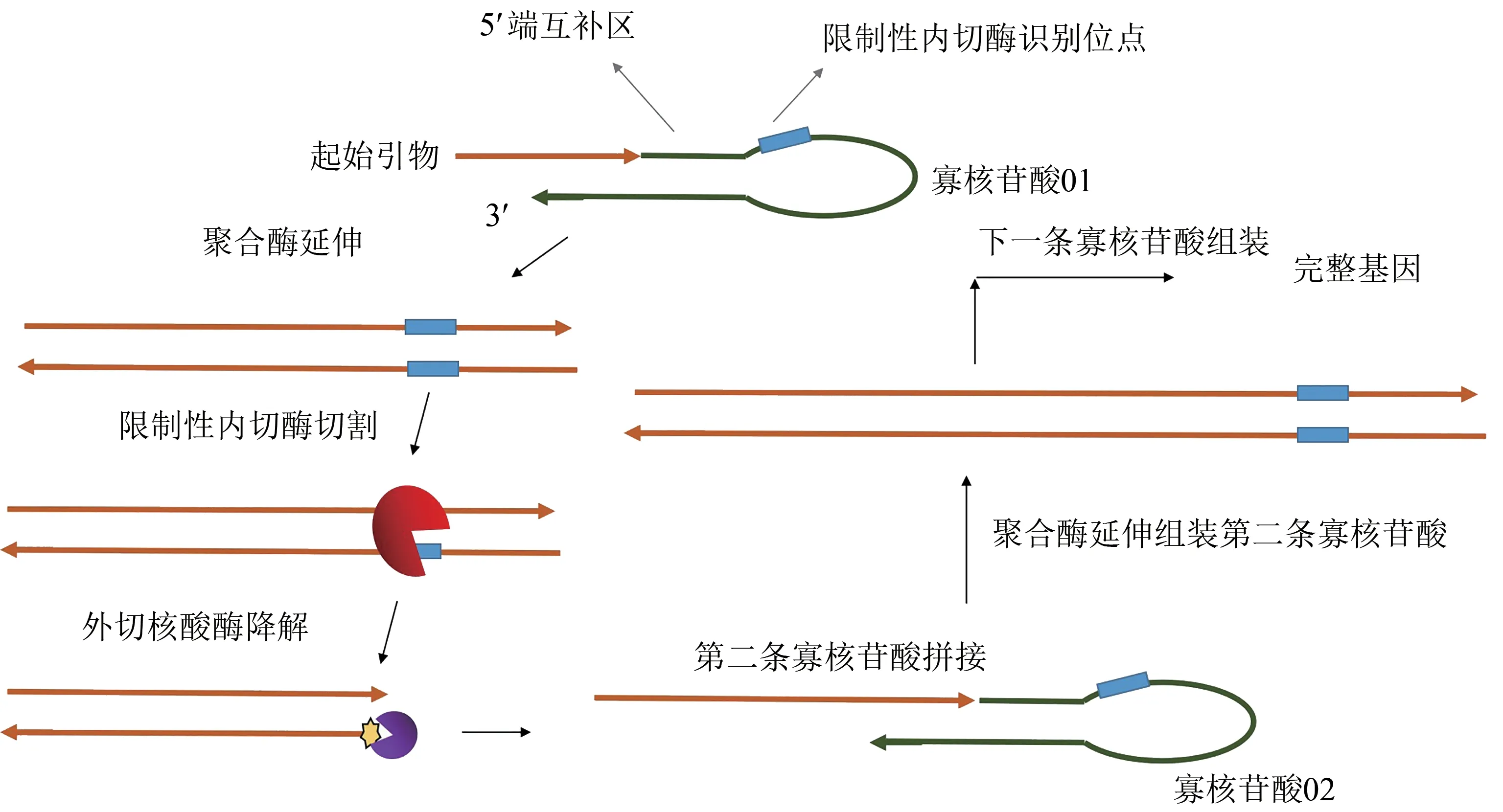
圖2 一種單向等溫的基因合成方法原理[54-55]Fig.2 A one-way isothermal gene synthesis method[54-55]
上述寡核苷酸拼裝方法,結(jié)合柱式寡核苷酸合成被大量應(yīng)用于工業(yè)化的基因合成生產(chǎn)中。然而,由于柱式合成寡核苷酸單堿基成本較高,使得基因合成的成本居高不下。而基于高通量芯片合成的寡核苷酸,其單條成本遠低于以往的柱式合成寡核苷酸,在降低基因合成成本上表現(xiàn)出很好的潛力。2004年,Tian等[57]首次提出一種基于芯片寡核苷酸的組裝方法。他們利用PCR擴增寡核苷酸池以提高用于基因拼裝的寡核苷酸的量,然后用反相互補的寡核苷酸雜交篩選來降低用于基因拼裝的寡核苷酸錯誤率,進一步地,利用一步酶組裝反應(yīng)將獲得的寡核苷酸片段組裝成多條基因。基于這種方法,他們成功合成組裝了21條編碼大腸桿菌30S核糖體亞基的基因。不同的研究在基因組裝規(guī)模、組裝長度、成本等多方面進行了優(yōu)化。這些成果幾乎都包括一個最核心的邏輯:通過設(shè)計的方法從一個混合文庫中抓取一條目的基因所需要的寡核苷酸片段進行組裝。比較有代表性的,2010年,Kosuri等[58]使用提前設(shè)計好的條形碼標記用于一組特定基因組裝的引物,通過先從合成文庫中擴增出組裝子庫,再從子庫中進一步擴增出用于基因組組裝的寡核苷酸片段的方式,實現(xiàn)了47個基因,總長35 kb基因的組裝。這個方法操作簡便,且能夠很好地進行擴展,具備工業(yè)生產(chǎn)的應(yīng)用潛力。2011年,Quan等[32]利用噴墨打印合成技術(shù),在一個芯片的微孔中,同時合成用于一個基因組裝的多條寡核苷酸,通過原位擴增和組裝,實現(xiàn)了多條基因在一個芯片上的合成。這項技術(shù)為開發(fā)工業(yè)化的高通量基因合成技術(shù)提供了很好的理論雛形,也促進了商業(yè)化技術(shù)平臺比如Twsit Bioscience噴墨打印合成平臺等的開發(fā)。另外,Kosuri領(lǐng)導(dǎo)的團隊,在2018年以及2020年分別發(fā)表了DropSynth 1.0[59]和DropSynth 2.0技術(shù)[60],利用微磁珠對同一個基因的不同引物進行富集,然后包裹在一個微液滴的酶反應(yīng)器內(nèi),通過擴增反應(yīng),實現(xiàn)基因的組裝。這種方法目前成功率還較低,但是為未來進一步降低高通量基因合成成本提供了可選的技術(shù)方案。
值得提及的是,雖然不同的技術(shù)都能夠?qū)崿F(xiàn)寡核苷酸的拼接,但是在工業(yè)化生產(chǎn)中,如何通過工藝優(yōu)化提升技術(shù)的穩(wěn)定性,以及降低技術(shù)的成本是關(guān)鍵。通過技術(shù)方法間的組合設(shè)計,開發(fā)低成本、高效、穩(wěn)定的工藝流程是一個可行的思路。另外,寡核苷酸的拼接常常面臨高GC、高AT和高重復(fù)序列合成困難的問題,尤其是對于依賴于PCA組裝或者包含PCA組裝流程的寡核苷酸拼接技術(shù)。由于高溫PCR擴增,涉及序列之間的退火再延伸的步驟,高GC/AT和高重復(fù)序列會增加不同寡核苷酸(或者DNA片段)之間的錯配,從而造成組裝失敗;同時高溫PCR擴增酶本身對于高GC或者高AT也會表現(xiàn)出一定程度的擴增困難。通過往PCR體系中添加GC擴增增強劑、優(yōu)化PCR程序以及在寡核苷酸設(shè)計時避免將這些區(qū)域包裹在重疊區(qū)內(nèi)等方式,能夠一定程度解決這些問題[61-62]。
2.2 基因合成糾錯與克隆篩選
用于基因合成拼裝的合成寡核苷酸常常伴隨著一定的錯誤,同時,基因合成拼接過程中,酶擴增或組裝也會引入一定概率的堿基錯誤,因此,為了減少寡核苷酸拼接中的堿基錯誤,一方面,可以通過優(yōu)化寡核苷酸拼接流程、程序以及選用高保真的擴增酶體系;另一方面,可以通過酶糾錯技術(shù)降低基因合成拼接過程的錯誤率。基于酶的糾錯技術(shù),主要是通過利用雙鏈DNA中錯誤序列和正確序列形成不匹配的區(qū)域,對錯誤序列進行識別或者切除。在基因合成過程中,由于酶擴增的產(chǎn)物是正確雙鏈DNA產(chǎn)物和含有錯誤的雙鏈DNA產(chǎn)物的混合物。通常需要先將產(chǎn)物進行變性再復(fù)性,然后通過錯配識別酶或錯配切割酶,將帶有錯配的雙鏈復(fù)合物去除。MutS是一種常用的錯配識別酶。在微生物體內(nèi),它能夠識別并結(jié)合多種錯誤的堿基以及單鏈的小環(huán)。利用MutS與錯配雙鏈的結(jié)合,然后利用合適的方法去除蛋白-雙鏈復(fù)合物,能夠降低基因組裝產(chǎn)物中的錯誤率[63-64]。將MutS固定在過濾柱上,也是一種能夠去除組裝基因中的錯誤鏈的辦法。2020年,徐健團隊[65]通過將MBP融合的MutS突變體固定在纖維素柱上,并利用該柱子識別并去除芯片寡核苷酸的錯誤,將基因合成組裝的堿基準確率提升了37.6倍。另外一些酶,則具備錯配識別并切斷錯誤配對雙鏈DNA的功能,包括T7 Endonuclease I、大腸桿菌Endonuclease V、米曲霉S1 Nuclease、芹菜CEL nuclease等[66]。其中一些酶被進一步拓展成為成熟的商業(yè)化產(chǎn)品,比如Transgenomic公司(美國)的Surveyor?內(nèi)切核酸酶,以及Thermo Fisher Scientific公司(美國)的CorrectASE內(nèi)切核酸酶(之前為美國Novici Biotech公司的ErrASE糾錯試劑盒)等。在多個已報道的基因合成組裝工作中,這些商品化的糾錯酶都表現(xiàn)出了一定程度的糾錯能力[32,58,67]。同時,關(guān)于不同的基因合成糾錯酶的糾錯能力比較分析表明,ErrASE能夠?qū)㈠e誤率降低到最低,而MutS能夠很好地增加正確的基因合成組裝數(shù)[68]。
盡管通過寡核苷酸拼接流程和體系優(yōu)化以及酶糾錯法能夠在一定程度上降低基因合成組裝的錯誤率,要獲得100%序列正確的基因仍然需要進一步的技術(shù)流程。利用大腸桿菌克隆篩選是一種常用的方法。具體流程包括:首先將寡核苷酸拼接產(chǎn)物或者經(jīng)過酶糾錯后的寡核苷酸拼接產(chǎn)物克隆到質(zhì)粒載體上,然后轉(zhuǎn)化至大腸桿菌,對經(jīng)抗性平板篩選的克隆進行培養(yǎng)并抽提質(zhì)粒,最后利用測序驗證篩選獲得包含有100%序列正確的質(zhì)粒。需要注意的是,在該步驟中,一些合成的基因克隆到質(zhì)粒載體后,在大腸桿菌中會發(fā)生不穩(wěn)定復(fù)制或產(chǎn)生毒性。這些基因常常因為克隆構(gòu)建困難或在大腸桿菌中易產(chǎn)生突變而造成基因合成的失敗。根據(jù)不同大腸桿菌菌株的遺傳特性,更換不同的菌株能夠在一定程度上緩解該問題。其中,Thermo Fisher Scientific公司推出了Stabl2和Stabl3大腸桿菌感受態(tài)系列,以及NEB公司推出了NEB Stable大腸桿菌感受態(tài)系列等用于解決基因克隆不穩(wěn)定的問題。同時,利用低拷貝的載體,也能夠?qū)崿F(xiàn)更穩(wěn)定的合成基因克隆。然而,在具體的基因合成應(yīng)用中,更換不同菌株或者載體克隆策略常常費時費力,且仍未能夠有效解決這類問題。因此,開發(fā)更加簡單通用的方法,比如不依賴于大腸桿菌克隆的方法,對于工業(yè)基因合成仍然尤為重要。
2.3 大片段基因合成組裝
由于寡核苷酸拼接組裝中的堿基仍存在一定的錯誤率,為減少首次克隆篩選獲得正確克隆的工作量,通常從寡核苷酸直接拼接組裝的基因長度會控制在3 kb以內(nèi)。對于更長的基因合成,則將首輪克隆篩選獲得的正確的基因片段組裝成更長的片段。一系列方法被應(yīng)用其中,如Golden Gate組裝[69-70]、Gibson組 裝[71]、 循 環(huán)LCR[72-73]、 雙 引 物TPA組裝[74]、BioBrick組裝[75]等。其中Golden Gate組裝法和Gibson組裝法,在大片段基因合成組裝應(yīng)用上相對成熟。基于Type IIS限制性內(nèi)切酶的Golden Gate克隆技術(shù),利用Type IIS限制性內(nèi)切酶在識別序列下游位置切割DNA的特點,能夠在任意需要相連的兩個片段末端創(chuàng)造互補配對的黏性末端[69-70]。這種方法能夠一次實現(xiàn)多個片段的組裝,也能通過多級組裝完成更多片段的組裝[76]。該組裝技術(shù)的優(yōu)點在于,操作簡便,能夠一次實現(xiàn)較長基因的組裝,同時對于包含各種特殊序列結(jié)構(gòu)的基因也能夠?qū)崿F(xiàn)很好的組裝效果。然而,如果人工合成的基因中有多個Golden Gate組裝使用的限制性內(nèi)切酶酶切位點,這個組裝方法失敗率較高。相比之下,Gibson組裝方法沒有酶切位點限制的問題,操作也很方便。利用片段之間互相重疊的同源區(qū)(通常20~150個堿基),在高溫聚合酶、高溫連接酶、外切核酸酶的作用下,能夠?qū)崿F(xiàn)多個片段在載體上的一步高效組裝。根據(jù)Gibson等[71]的測試結(jié)果,其能夠有效組裝DNA長度達到數(shù)十萬堿基。然而缺點是,當(dāng)片段之間重疊區(qū)GC或者AT含量很高時,可能發(fā)生一定的錯誤。結(jié)合限制性內(nèi)切酶切割,修改的Gibson組裝方法能夠一定程度解決這一問題[77]。另外,依賴于酵母同源重組系統(tǒng),也能夠很好地實現(xiàn)多個帶有同源互補區(qū)設(shè)計的片段的一步組裝[78-80]。對于一些在大腸桿菌組裝克隆中有困難的基因合成,酵母同源重組組裝是一個很好的選擇。然而,由于目前常用的一些質(zhì)粒載體不帶有酵母的復(fù)制系統(tǒng),且基于酵母體系的質(zhì)粒制備成本相對較高,一定程度上限制了該方法的應(yīng)用。
由于越大的質(zhì)粒,在大腸桿菌中高拷貝復(fù)制時越不穩(wěn)定。在大腸桿菌中組裝的基因片段長度通常小于15 kb。對于更長的基因,可以利用特殊的克隆載體,比如包含單拷貝F質(zhì)粒復(fù)制因子的BAC或者改造的BAC載體[81-82],來進行組裝。然而,這類質(zhì)粒克隆效率低下,規(guī)模化操作困難。因此,一些超大片段的組裝可以通過轉(zhuǎn)化至酵母載體上,在酵母中完成[78-80]。盡管目前的技術(shù)研究已經(jīng)能夠?qū)崿F(xiàn)長度達到數(shù)十萬堿基的片段組裝[71],但目前大多商業(yè)化交付的基因合成仍然局限在15 kb以內(nèi)。因此,如何建立更加高效、標準化的十萬堿基級別的大片段基因組裝方法及工藝,實現(xiàn)規(guī)模化的大片段基因合成交付,對于促進更廣泛、深入的合成生物學(xué)研究及應(yīng)用有著革命性的意義和價值。
3 基因組合成技術(shù)
合成組裝一個生物體完整的基因組并使之有生命活性,一直是DNA合成領(lǐng)域努力的方向之一。2002年,Cello等[83]利用化學(xué)合成的寡核苷酸原料,從頭合成了帶T7 RNA聚合酶啟動子的脊髓灰質(zhì)炎病毒cDNA,并基于合成的cDNA,轉(zhuǎn)錄出病毒的RNA,同時在Hela細胞提取液中組裝出有活性的病毒顆粒。它利用平均長度在69個堿基的合成寡核苷酸原料,首先通過末端互補方式,拼接成400~600個堿基的基因片段;然后將這些片段插入到質(zhì)粒載體中,經(jīng)測序驗證后,通過逐步克隆的方式拼接成完整的病毒cDNA。緊接著,2003年,Venter實驗室[84]利用優(yōu)化的方法合成了一個5386 bp的細菌噬菌體?X174。該團隊利用平均長度在42個堿基的寡核苷酸,首先進行PAGE純化,然后利用T4多聚核苷酸激酶磷酸化,在Taq連接酶的作用下,將寡核苷酸連接成主要大小在700 bp左右的片段混合物;進一步地,通過PCA組裝將這些片段混合物組裝成完整的噬菌體DNA,經(jīng)酶切環(huán)化后,轉(zhuǎn)入大腸桿菌細胞中獲得侵染能力的噬菌體。這兩個工作開啟了基因組合成的新紀元。
隨著基因合成技術(shù)的進步,目前拼裝一個完整的病毒或者噬菌體基因片段變得容易得多,甚至可以由CRO服務(wù)公司單獨完成。2020年,Thao等[85]利用在第三方服務(wù)公司合成的基因片段,基于酵母組裝平臺,在T7 RNA聚合酶轉(zhuǎn)錄作用下,在一周內(nèi)就完成了2020年新型冠狀病毒SARS-CoV-2的合成。相對于幾千堿基到數(shù)萬堿基病毒或者噬菌體基因組,超過Mb水平的細菌基因組和酵母因組要大得多,因此,合成也困難得多。近十年來,多個研究成果實現(xiàn)了不同細菌基因組或者酵母染色體合成[86-92]。這些研究在基因組合成設(shè)計、基因組大片段拼接與分離、合成糾錯調(diào)試、功能篩選與測試等多個關(guān)鍵技術(shù)環(huán)節(jié)都進行了不同程度的優(yōu)化與測試。究其核心技術(shù)邏輯,可以大致概括為“一次從頭合成”和“逐步替換從頭合成”兩種。其中,“一次從頭合成”的代表工作為2010年,Gibson等[87]從寡核苷酸合成了絲狀支原體Mycoplasma mycoides的基因組。在這個工作中,他們首先利用將從供應(yīng)商獲得的基于化學(xué)合成寡核苷酸組裝的1078個1080 bp片段,利用酵母同源重組組裝成109個10 080 bp的組裝產(chǎn)物;然后基于這些10 080 bp的片段,利用酵母同源重組進一步組裝獲得11個100 kb的組裝片段;進一步基于這些大片段,利用酵母組裝成為1 077 947 bp完整的基因組。隨后,完整的基因組合成被轉(zhuǎn)移到另外一個支原體受體細胞中,在篩選標記的協(xié)助下獲得有功能的細胞。“逐步替換從頭合成”是“國際Sc2.0酵母基因組合成計劃”采用的合成方法。其特點是,先利用常規(guī)的基因合成拼接技術(shù),從寡核苷酸逐級拼裝出10 kb的基因片段。然后將多個10 kb的基因片段經(jīng)酵母同源重組系統(tǒng)拼接成30~60 kb的大片段,同時在篩選標記的協(xié)助下替換野生型基因組中對應(yīng)序列的片段。通過30~60 kb大片段逐步替換的方式,實現(xiàn)人工酵母染色體的合成。目前,“國際Sc2.0酵母基因組合成計劃”已經(jīng)基于該方法,發(fā)表了6.5條染色體的合成工作[89-90]。相對于“一次從頭合成”,這個方法的好處是,當(dāng)出現(xiàn)設(shè)計的合成替換片段對酵母功能造成極大影響時,能夠調(diào)整設(shè)計策略,進行及時的糾錯修正。
盡管目前細菌、酵母染色體已合成成功,但對于更大的動植物細胞基因組合成仍然面臨諸多挑戰(zhàn)。比如,Mb級別的基因組片段的快速合成及其在動植物細胞基因組的高效替換被認為是動植物基因組合成的技術(shù)策略之一,然而也是需要進一步開發(fā)突破的關(guān)鍵技術(shù)瓶頸。同時,超大基因組的合成也面臨著諸多問題,包括:①合成基因組大片段的堿基突變、缺失、移位等,需要大量的糾錯測試工作;②部分大片段的質(zhì)粒轉(zhuǎn)化及組裝可能面臨失敗,需要返工;③保證合成的基因組產(chǎn)生功能,需要一定的前期調(diào)研與設(shè)計工作。生命體的復(fù)雜性,使得生命再造的過程充滿了藝術(shù)性和不確定性;然而,正是因為這種不確定性,每一步科學(xué)上的跨越才會無比珍貴。隨著合成和組裝DNA技術(shù)的不斷進步,自動化儀器平臺商業(yè)化的日趨成熟,我們有理由相信未來實現(xiàn)一個基因組的合成和今天的基因組測序一樣簡單、高效。
4 新一代酶法DNA合成技術(shù)
合成生物學(xué)的高速發(fā)展催生了大量的基因合成需求。傳統(tǒng)的化學(xué)寡核苷酸合成法逐漸凸顯出其缺點:①合成長度太短,且堿基錯誤率高,導(dǎo)致合成拼裝過程耗時耗力;②其工藝過程要求高,通常需要控制在一個無水無氧的反應(yīng)環(huán)境內(nèi);③合成過程中產(chǎn)生大量的污染性有機化學(xué)廢棄物,對環(huán)境不友好。因此,基于生物酶催化的DNA合成技術(shù)給合成生物學(xué)家們帶來了新的曙光。
基于模板DNA的聚合酶延伸反應(yīng)技術(shù)已經(jīng)被廣泛應(yīng)用于DNA的復(fù)制與擴增,如PCR技術(shù)。利用可逆化學(xué)修飾堿基的聚合酶延伸反應(yīng)[93],“邊合成邊測序技術(shù)”(SBS,sequencing by synthesis)已經(jīng)很好地應(yīng)用于高通量DNA測序。這給生物酶催化的DNA合成提供了重要的理論啟示。近十幾年來,基于末端轉(zhuǎn)移酶(terminal deoxyribonucleotidyl transferase,TdT)的新一代酶法DNA合成技術(shù)越來越受到重視。1959年,Bollum[94-95]首次闡述末端轉(zhuǎn)移酶TdT可以用于不依賴于模板的DNA合成。1962年,Bollum發(fā)現(xiàn)將起始堿基的3′-OH基團封閉掉,能夠阻止末端轉(zhuǎn)移酶的堿基聚合反應(yīng),證實了dNTP是加到起始堿基的3′-OH端。與此同時,Bollum[96]也概念性地提出,能夠利用封閉3′-OH端的堿基單體,基于TdT末端轉(zhuǎn)移酶,合成核苷酸多聚體。在分類學(xué)上,TdT末端轉(zhuǎn)移酶隸屬于聚合酶X家族的一部分。在生物體內(nèi),它的主要功能是通過隨機的堿基插入,增加抗原受體多樣性[97-98]。2008年,Ud-Dean等[99]進一步完善了基于TdT末端轉(zhuǎn)移酶的理論模型:利用不依賴于模板的TdT末端轉(zhuǎn)移酶堿基聚合反應(yīng),基于可逆化學(xué)修飾堿基,根據(jù)任意設(shè)計的序列DNA,能夠?qū)崿F(xiàn)不依賴于模板的長片段人工DNA的合成。
基于TdT酶法DNA合成技術(shù),從理論走向?qū)嶋H應(yīng)用的一個關(guān)鍵點是,需要獲得一個高效的TdT酶和可逆修飾堿基組合。由于TdT酶可以一次延伸多個連續(xù)的堿基,為了保證TdT酶依賴的DNA合成按照預(yù)定的序列一個個延伸,需要對堿基單體進行修飾,以保證第n+1個堿基合成完成后,反應(yīng)能夠被終止,而不會繼續(xù)進行n+2個堿基的反應(yīng)。同時堿基的修飾基團能夠通過化學(xué)、物理或者生物的方法切除,以繼續(xù)下一個堿基的合成。3′-O修飾的可逆dNTPs是TdT酶依賴的單鏈DNA合成反應(yīng)很好的底物(圖3)。2016年Mathews[100]報道了合成的NB-dNTPs[3′-O(-2-nitrobenzyl)-2′-deoxyribonu‐cleoside triphosphates] 以 及DMNB-dNTPs[3′-O-(4,5-dimethoxy-2-nitrobenzyl)-2′-deoxyribonucleoside triphosphates]能夠被TdT酶催化利用,同時能夠阻斷TdT酶的第n+2延伸反應(yīng)。同時,NB基團及DMNB基團能夠在紫外線的作用下實現(xiàn)完全的降解。此外,可逆的修飾基團也可以加在堿基上面[101],但是實際的應(yīng)用效果未見充分的報道。
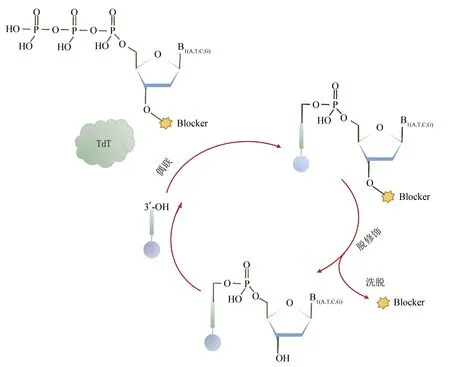
圖3 基于3′-O修飾的可逆dNTPs TdT酶法DNA合成示例[99-100]Fig.3 Illustration of TdTenzymatic DNA synthesis based on 3′-O modified reversible dNTPs[99-100]
另外,2018年,Keasling實驗室[102]報道了一種新的策略,將dNTPs利用一個可以被切割的間臂偶聯(lián)在TdT酶催化中心附近的位置,當(dāng)TdT酶催化dNTPs進入單鏈寡核苷酸以后,TdT酶仍然結(jié)合在單鏈寡核苷酸上,阻止下一個TdT-dNTPs進入反應(yīng)。通過切割間臂,在釋放TdT酶后,新的TdT-dNTPs可以繼續(xù)進入反應(yīng)。雖然TdT-dNTPs目前使用成本價格較高,但是該方法反應(yīng)速度快,同時具備很好的優(yōu)化升級潛力,為TdT酶介導(dǎo)的DNA合成技術(shù)開發(fā)提供了很好的可選策略。
盡管目前的研究表明TdT酶能夠很好地應(yīng)用于
單鏈DNA的合成,然而開發(fā)可大規(guī)模交付且成熟的單鏈DNA合成技術(shù)仍需投入大量的研究,包括:通過TdT酶工程改造,提升其針對可逆修飾堿基的活性;整體優(yōu)化酶循環(huán)反應(yīng)工藝,提升每輪反應(yīng)的堿基合成效率等。目前,至少有4家公司專注于這個方面的技術(shù)開發(fā),包括DNAScript(法國)、Nuclera(英國)、Molecular Assemblies(美國)和Ansa Biotechnologies(美國)等。這些公司已獲得了大量的資金支持,同時,已經(jīng)基于生物酶法實現(xiàn)了一定長度的單鏈寡核苷酸的合成,甚至開發(fā)出了基于酶合成法的合成儀原型。另外,值得提及的是,在技術(shù)未成熟之前,嘗試更多的方案對于實現(xiàn)生物酶法DNA合成技術(shù)的工業(yè)化應(yīng)用仍然具有重要價值,比如不依賴模板的其他聚合酶[103-104]、RNA連接酶[105-106]等。2019年,Halpain等[104]就報道了一種方法,基于DNA聚合酶,借助于瞬時的寡核苷酸雜交,也能夠?qū)崿F(xiàn)單鏈DNA的合成。
5 DNA合成與DNA數(shù)據(jù)存儲
DNA合成作為DNA數(shù)據(jù)存儲的關(guān)鍵技術(shù)基礎(chǔ),是DNA數(shù)據(jù)存儲從理論走向應(yīng)用的基石。2012年,Church等[3]利用合成的54 898條159 nt寡核苷酸庫存儲了53 426個單詞、11張JPG圖片和一個JavaScript程序。每條寡核苷酸包含了96 nt的數(shù)據(jù)區(qū),左右擴增引物區(qū)和測序引物區(qū)各22 nt以及一個19 nt的信息地址(索引)區(qū)。2017年,Erlich等[4]利用72 000條200 nt的寡核苷酸庫,存儲了2 146 816 bytes的信息。每一條寡核苷酸包含有128 nt的數(shù)據(jù)區(qū),16 nt的種子區(qū)(類似于索引堿基功能),8 nt的糾錯區(qū),以及24 nt的左右擴增引物區(qū)。2018年,Organick等[5]利用9個合成的寡核苷酸文庫,總計約1300萬條寡核苷酸,存儲了200 MB的數(shù)據(jù)信息。此外,也有研究工作利用寡核苷酸拼裝的基因片段[107]、克隆的質(zhì)粒[108-109]等實現(xiàn)數(shù)據(jù)信息在DNA中的存儲。然而,鑒于成本以及可擴展性等原因,目前DNA數(shù)據(jù)存儲技術(shù)主要依賴合成的芯片寡核苷酸文庫。DNA合成的長度、成本及速度是影響DNA數(shù)據(jù)存儲應(yīng)用的關(guān)鍵因素。
存儲數(shù)據(jù)信息的DNA長度直接決定存儲信息堿基利用率。由于芯片寡核苷酸合成長度有限,在存儲數(shù)據(jù)信息時,需要將二進制信息編碼的寡核苷酸序列,拆分成一系列的序列片段。為了在測序解讀時,能夠?qū)⑦@些序列片段拼接成完整的存有二進制比特信息的DNA序列,需要在這些序列片段上添加索引序列(或者位置序列)[3,5],用來標識這些序列片段對應(yīng)于存有二進制比特信息的DNA序列中的位置;比如用4個堿基代表256個序列片段的索引位置,AAAA=1,AAAT=2…GGGG=256。當(dāng)拆分序列片段越多時候,用來標識片段位置的索引序列的長度就越長(通常,拆分序列的個數(shù)m≤4n,n為索引序列的堿基數(shù))。另外,為了方便信息的隨機讀取和備份,還需要在存儲信息的序列兩端添加擴增引物[3-5]。甚至,還需要根據(jù)需求添加糾錯碼等[110]。在這種情況下,用于存儲數(shù)據(jù)信息的寡核苷酸長度越長,存儲信息堿基利用率就越高。例如,若采用Church等[3]的方法存儲數(shù)據(jù)信息,當(dāng)合成寡核苷酸長度達到600 nt時,存儲信息堿基利用率能夠從63%提升到接近90%的水平(圖4)。盡管,寡核苷酸拼接的更長的基因片段也可以用于數(shù)據(jù)存儲,然而由于單堿基的成本將增加10~100倍,推廣使用困難。因此,未來的技術(shù)研究可以更加關(guān)注低成本長片段芯片寡核苷酸合成技術(shù)開發(fā)。
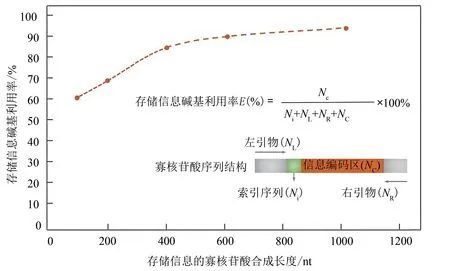
圖4 存儲信息堿基利用率與寡核苷酸合成長度之間的關(guān)系[本圖根據(jù)Church等[3]使用的方法,左右引物各22 nt,索引(地址)序列19 nt,假定合成長度不同的情況下,計算存儲信息堿基利用率]Fig.4 Relationship between base utilization of data storage and oligo length[Referring to methods used by George Church et al.,base utilization of data storage is calculated given that oligo length is different,while both the left and right flanking primers are 22 nt,and the index(address)sequence is 19 nt]
目前,DNA數(shù)據(jù)存儲的主要成本來源于DNA合成。即便是最便宜的芯片合成寡核苷酸,其單堿基合成成本仍高于測序成本近5個數(shù)量級。按照當(dāng)前DNA合成成本計算,仍需降低6~8個數(shù)量級才能使得DNA數(shù)據(jù)存儲成本與目前硬盤存儲的(約100元/TB)相近。然而,不同于其他DNA合成應(yīng)用,用于DNA數(shù)據(jù)存儲的合成DNA能夠在一定程度上降低對保真度的要求。由于目前合成技術(shù)一次合成的DNA分子數(shù)通常大于fmol級別,使得即便在一定的堿基錯誤率情況下,合成DNA中針對一條序列也有不同的分子用于信息糾錯。同時,經(jīng)特殊設(shè)計的數(shù)據(jù)冗余及糾錯碼,能夠進一步提升低保真合成DNA中存儲數(shù)據(jù)信息的解讀準確性[5-6,110]。DNA合成保真度要求的降低,一定程度降低了DNA合成工藝的要求,從而有利于開發(fā)出更低成本的DNA合成技術(shù)。如依賴未修飾dNTPs的TdT酶法合成已被驗證其在DNA數(shù)據(jù)存儲中的應(yīng)用[111]。未來在該方向的研究將有望進一步開發(fā)適配于DNA數(shù)據(jù)存儲的低成本合成技術(shù)。值得提及的是,利用通用合成的DNA片段,基于類似于“活字印刷”的原理來存儲數(shù)據(jù)信息,也可能是一種非常有效的降成本方式。比如將英文的26個字母分別存儲在通用合成DNA上,然后通過酶拼接或者其他方法,在存儲信息時,進行自由組合,進而反復(fù)使用一次合成的DNA分子,能夠潛在地降低成本,然而這些方法仍然需要進一步的開發(fā)。
此外,DNA合成的速度也決定了DNA信息存儲的寫入速度。目前的DNA合成技術(shù)依賴循環(huán)的化學(xué)反應(yīng)或者酶催化反應(yīng),而每輪化學(xué)反應(yīng)或者酶反應(yīng)都需要較長的時間。如亞磷酰胺化學(xué)DNA合成,基于自動化合成儀的每輪化學(xué)合成反應(yīng)耗時在數(shù)分鐘到十幾分鐘。換言之,以合成200 nt的寡核苷酸為例,其耗時將達到幾十個小時。相比于硬盤存儲的快速寫入,這個速度仍相距甚遠。因此,除降低合成DNA的成本外,合成速度的提升也是DNA數(shù)據(jù)存儲應(yīng)用實現(xiàn)的關(guān)鍵。
同時,DNA合成作為DNA數(shù)據(jù)存儲技術(shù)流程中的一個重要環(huán)節(jié),可以與其他環(huán)節(jié)的技術(shù)工藝進行整合優(yōu)化,以實現(xiàn)DNA數(shù)據(jù)存儲整體成本的降低與效率的提升。如在信息的編碼環(huán)節(jié),通過提升從二進制信息編碼獲得的DNA序列的GC均一度,來提升DNA合成技術(shù)環(huán)節(jié)的成功率;通過開發(fā)適用于合成和測序錯誤率的糾錯編碼技術(shù),提升在測序及后續(xù)數(shù)據(jù)解讀過程中的準確性等。另外,為了實現(xiàn)更好的信息解讀,還需要進一步地提升測序技術(shù)以及數(shù)據(jù)解讀技術(shù)。比如,可以通過縮短DNA測序流程的建庫時間以及整合快速的堿基序列讀取技術(shù),實現(xiàn)存儲數(shù)據(jù)DNA的實時、快速讀取;通過開發(fā)測序信號讀取與解碼一體化算法,加速A/T/C/G信息到0/1二進制信息的讀取過程等。最終,通過開發(fā)基于數(shù)據(jù)編碼、DNA合成、DNA測序、數(shù)據(jù)讀取的一體的高效技術(shù)流程,實現(xiàn)DNA數(shù)據(jù)存儲的大規(guī)模應(yīng)用。
6 總結(jié)和展望
如果說DNA測序技術(shù)打開了人類對生命遺傳規(guī)律的認知之門,那么人工DNA合成技術(shù)使人類進一步深度認知、改造甚至創(chuàng)造生命成為可能。DNA合成技術(shù)的發(fā)展使得生命科學(xué)從測序帶來的可觀測、可理解、可描述的數(shù)字化生命時代向可預(yù)測、可定量、可創(chuàng)造的合成生物學(xué)工程化時代邁進。
經(jīng)過將近70年的發(fā)展,DNA合成技術(shù)已經(jīng)從若干個寡核苷酸堿基的合成跨越到Mb級微生物基因組的合成(圖5)。大規(guī)模的寡核苷酸、基因的合成已經(jīng)實現(xiàn)了商品化交付,同時也很好地服務(wù)于科研及生物技術(shù)產(chǎn)業(yè)的發(fā)展。目前,高通量芯片DNA合成技術(shù)的單次合成堿基量超Mb,且成本僅為柱式合成的1/10 000到1/100。同時,基于芯片技術(shù)的更高通量的DNA合成技術(shù),及其原位基因合成組裝技術(shù)也正逐漸走向商業(yè)化。與此同時,新一代酶法DNA合成技術(shù)也為未來的更高通量及更低成本DNA合成帶來了曙光。DNA合成儀器制造公司以及DNA合成服務(wù)公司也獲得了眾多產(chǎn)業(yè)化發(fā)展機遇。然而,相比于測序技術(shù),DNA合成技術(shù)仍處于較早期水平。
目前DNA合成成本仍然較高,極大限制其在DNA數(shù)據(jù)存儲等合成生物學(xué)領(lǐng)域的應(yīng)用。因此,降低DNA合成成本仍是未來技術(shù)開發(fā)的關(guān)鍵。除開發(fā)核心技術(shù)流程外,適當(dāng)降低用于DNA合成的化學(xué)和生物試劑原料的成本等也是影響DNA合成成本的重要因素。此外,人工成本也是DNA合成中的重要組成部分,尤其是高耗時耗力的基因和基因組合成、拼裝過程。開發(fā)高集成的自動化平臺對于降低人工成本、提高合成效率將起到重要的作用。
盡管目前已實現(xiàn)微生物基因組的從頭合成,但對于102kb級以上大片段DNA的合成,其合成周期仍相對較長,達數(shù)月之久,且失敗率較高。同時,目前基因組合成仍然停留在微生物水平,對于動植物基因組的合成仍需要突破眾多技術(shù)瓶頸。這一定程度上限制了DNA合成在DNA數(shù)據(jù)存儲、合成生物學(xué)生命再造等領(lǐng)域的應(yīng)用。開發(fā)更高效的基因組水平的大片段DNA合成技術(shù),將是DNA合成從生物體局部基因改造到大規(guī)模全局生命再造應(yīng)用的關(guān)鍵。另外,盡管新一代酶法DNA合成技術(shù)通過最近十幾年的發(fā)展,擁有一定的技術(shù)基礎(chǔ),距離工業(yè)規(guī)模的合成交付仍然有一定的距離,需投入更多的創(chuàng)新研究和持續(xù)的努力。
DNA合成技術(shù)的發(fā)展促進了代謝工程改造[112]、酶工程改造[113]、抗體工程[114]、IVD診斷[115]、寡核苷酸藥物[14]、DNA數(shù)據(jù)存儲[1,3-6]等多個合成生物學(xué)領(lǐng)域的發(fā)展。尤其是面向特定應(yīng)用的DNA合成技術(shù),將會給下游應(yīng)用領(lǐng)域帶來革命性的變革。比如通過建立DNA合成設(shè)計到特定應(yīng)用的快速自動化合成平臺,將加速有益于人類功能活性物質(zhì)的生產(chǎn)或藥物分子的菌株改造效率[116-120]。作為DNA數(shù)據(jù)存儲流程的基礎(chǔ)技術(shù),人工DNA合成技術(shù)是DNA數(shù)據(jù)存儲從概念走向大規(guī)模應(yīng)用的關(guān)鍵。在全球數(shù)據(jù)大爆發(fā)的背景下,開發(fā)針對DNA數(shù)據(jù)存儲的長片段、低成本、快寫入的DNA合成技術(shù)(圖5), 對于加速DNA數(shù)據(jù)存儲的應(yīng)用以及解決人類面臨的數(shù)據(jù)危機尤為重要。
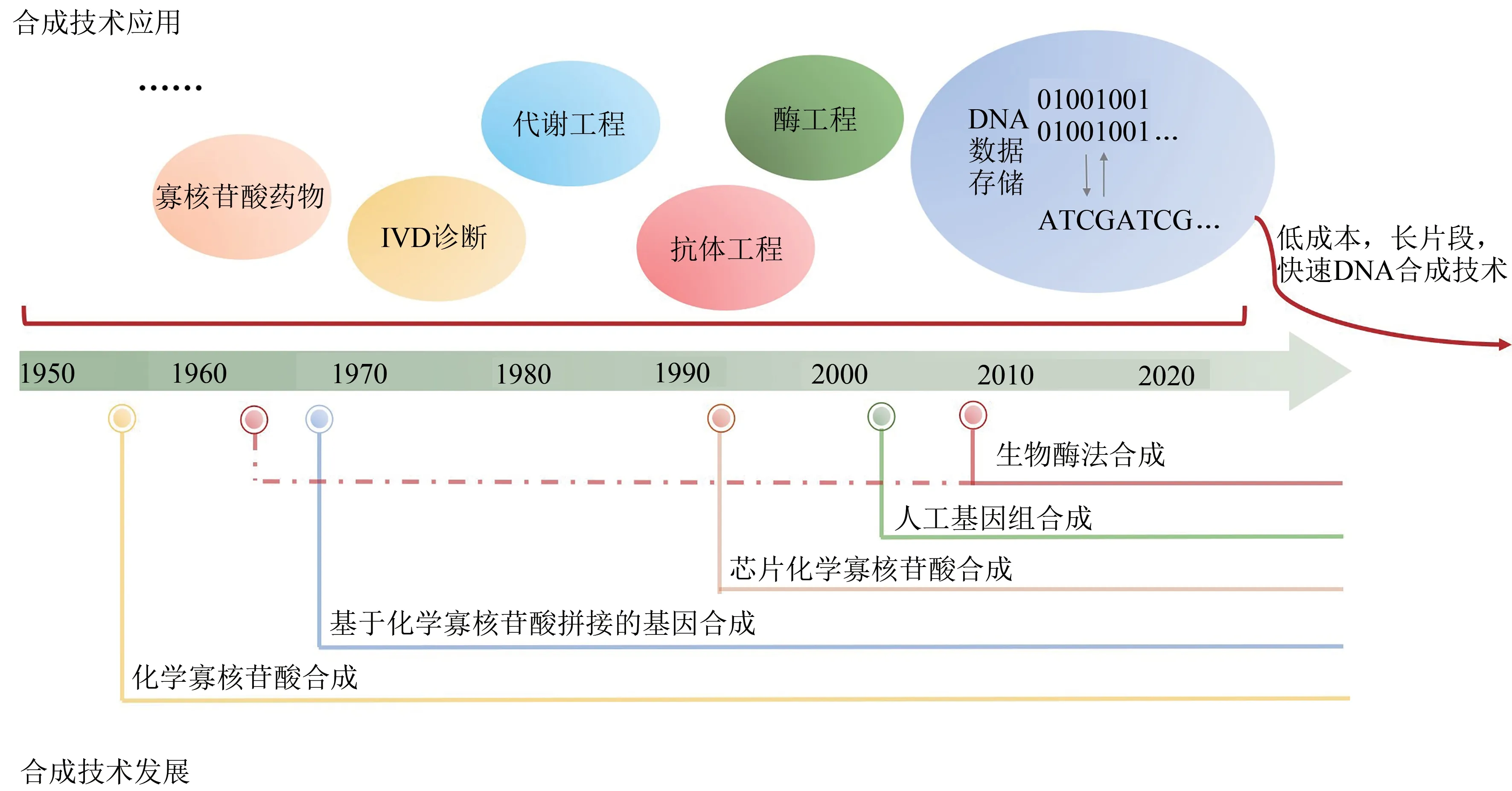
圖5 DNA合成技術(shù)發(fā)展與應(yīng)用Fig.5 Development and application of DNA synthesis technology
致謝:感謝中國科學(xué)院深圳先進技術(shù)研究院合成生物學(xué)研究所葉健文副研究員對本論文提出的修改建議。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學(xué)生數(shù)理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
意林原創(chuàng)版(2016年10期)2016-11-25 10:28:30
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
