干旱條件下棉花根際真菌多樣性分析
2021-07-19 09:43:12岳丹丹AbidUllah張獻龍楊細燕
作物學報 2021年9期
關鍵詞:植物
岳丹丹 韓 貝 Abid Ullah 張獻龍 楊細燕
研究簡報
干旱條件下棉花根際真菌多樣性分析
岳丹丹 韓 貝 Abid Ullah 張獻龍 楊細燕*
華中農業大學作物遺傳改良國家重點實驗室, 湖北武漢 430070
植物根際微生物群落對植物生長和適逆性至關重要, 本研究對干旱條件下棉花根際真菌群落進行分析, 旨在探明干旱脅迫對棉花根際真菌多樣性和群落結構的影響, 為利用有益微生物提高棉花水分利用率提供理論依據。以陸地棉Jin 668 (cv. Jin668)為試驗材料, 采用盆栽控水方式, 對處于開花期的棉花根際土壤(SDP)和未種植棉花土壤(SOPD)進行干旱處理, 正常澆水的棉花根際土壤(SPN)和無棉花土壤(SNPN)為對照。從中采集土壤樣品, 提取DNA, 采用Illumina Miseq對真菌ITS1區域進行高通量測序, 研究土壤中真菌多樣性。結果共鑒定到970個OTUs, SNPN、SOPD、SPN和SDP樣品中真菌OTUs數量分別為481、528、743和752個, 其中288個OTUs為所有組共有。對獲得OTUs進行門、綱、目、科和屬5個分類水平的劃分表明, 棉花根際真菌群落結構主要由子囊菌門(82.70%)和擔子菌門(10.15%)組成; 干旱處理使糞殼菌綱(Sordariomycetes)、糞殼菌目(Sordariales)和毛殼菌科(Chaetomiaceae)豐度顯著降低, 而散囊菌目(Eurotiales)、發菌科(Trichocomaceae)、曲霉屬()和青霉屬()的豐度顯著增加。多樣性分析結果顯示, 與未種棉花的土壤相比, 有棉花的土壤中真菌群落的α多樣性顯著增加; 同時, SPN和SDP之間的真菌群落結構更相似, 而與SNPN和SOPD間差異較大。研究表明, 棉花根際存在豐富的真菌群落, 干旱對土壤中真菌的豐度和多樣性有顯著影響。本研究從微生物的角度為提高棉花耐旱性的研究提供新見解。
棉花; 干旱; 真菌群落多樣性; 根際微生物; 高通量測序
干旱是世界糧食安全和植物生產力的主要制約因素之一, 對農作物生長、發育和農藝性狀等產生不利影響[1]。棉花是主要經濟作物之一, 表現出比其他主要作物如小麥、水稻和玉米對干旱條件更強的耐受性[2]。然而, 干旱等極端環境條件仍然會限制棉花的生長和產量、影響其纖維品質。在長期的進化過程中, 棉花已經形成了一系列形態、生理、生化和分子策略應對干旱脅迫[3]。根系是吸收養分和水分的主要部位, 同時也是分泌排泄物質的重要部位, 植物根系分泌物較多, 營養豐富、微生物多樣性豐富[4]。植物影響著其周圍及體內微生物的群落結構, 這些微生物又通過其生命活動影響植物的生長發育。微生物組被認為是植物的第2套基因組, 對植物的生長和適逆性至關重要[5]。
研究表明, 特定的根際和內生微生物有助于提高棉花對大麗花輪枝孢菌的抗性[6]。除了保護植物免受生物和非生物脅迫的內在系統外, 在遭受干旱脅迫時, 植物可以與相互作用的微生物(特別是對土壤變化迅速響應的各種土壤微生物群落)建立有益聯系, 從而減輕干旱脅迫損傷[7-8], 如來自谷子的耐干旱根際細菌可以在干旱脅迫條件下促進植物生長[9]。同時, 一些體外試驗也表明, 根系微生物在植物抵御鹽堿、干旱和抗病等非生物脅迫過程中發揮作用[10-12]。水稻[13]、玉米[14]、擬南芥[15-16]等多種植物的根系微生物與宿主植物適逆能力相關性的研究也表明有益微生物能提高植物對逆境的適應性。
進一步研究表明, 微生物能合成植物生長調節因子以減輕脅迫對寄主植物的不利影響, 如叢枝菌根真菌可能通過影響宿主植物生長素、細胞分裂素和乙烯等激素分泌, 進而導致植物根系形態發生變化, 增強植物的抗旱性[17]; 水楊酸、茉莉酸、乙烯等植物激素介導的信號通路調控根際促植物生長細菌誘導植物產生對病原菌的免疫反應, 即誘導系統抗病性, 增強植物的抗病性[18]; 基于植物和真菌中赤霉素(gibberellic acids, GA)生物合成途徑也被闡明[19]。此外, 微生物還會改變土壤酸堿度、土壤結構、土壤肥力和氧氣利用率[20], 如叢枝菌根真菌可通過改善根系形態和改良土壤結構來提高植物的抗旱性[17]。微生物的這些特性可抑制植物病原體并增強植物對高溫、鹽、干旱和金屬毒性的耐受性[21]。同時, 微生物在植物離子轉運和營養吸收方面也發揮著重要作用, 如叢枝菌根真菌可以通過改變宿主植株的根系形態和菌絲網絡的形成, 以擴大植株對土壤養分吸收范圍; 植物釋放的有機酸、磷酸酶和質子等根系分泌物可以改變土壤結構和理化性質, 與根際微生物共同作用溶解土壤中難溶的礦質元素(如P和Fe), 提高植株對離子的轉運能力、促進其吸收[22]。其次, 微生物還能調控宿主植物的水通道蛋白基因、干旱相關基因以促進植物在干旱脅迫下的生長和代謝[23], 如叢枝菌根真菌能夠特異性誘導苜蓿()水通道蛋白基因的表達, 進而調節苜蓿對硝態氮或銨態氮的吸收[24], 或通過誘導相關磷轉運蛋白基因的特異性表達等多種方式, 為植物提供生長和抗逆所需的礦質元素[22,25]。
反過來, 根際微生物群落結構變化也受植物的影響, 如利用高通量測序技術測定的花鈴期土壤中細菌群落結構結果表明, 黃萎病抗病品種在花鈴期的細菌群落結構優于感病品種[12]。此外, 根際微生物的群落結構也受其他多種因素影響。在不同的發育階段, 棉花根際細菌的群落結構變化很大[26]; 土壤微生物群落對長期連作棉花也有相似的反應, 即在相同的生境中, 微生物群落表現出相似性[27]; 相對無植物種植的土壤, 即使在干旱脅迫下棉花根際也有著豐富的細菌群落, 這可能有助于提高植物的耐旱性[28]。
植物-微生物互作為研究植物適逆性提供了一個新視角, 對有益微生物的使用將有助于農業生產系統走向一個更具生產力的時代。目前, 高通量測序方法為根際微生物群落的研究提供了新的思路和方法。本研究利用高通量測序技術, 通過對正常和干旱條件下棉花根際真菌群落特征進行分析, 研究干旱對棉田土壤中真菌多樣性和群落結構的影響, 確定最適合棉花抗旱研究的真菌種類, 有助于挖掘棉田中有益微生物, 同時對提高棉花耐旱性的研究具有重要的理論意義。
1 材料與方法
1.1 植物材料、干旱處理及樣品采集
土壤的收集、材料的種植及干旱處理參照文獻[28], 從長期連作棉花試驗田采集土壤, 將土壤在陽光下干燥1個月, 然后用2 mm的篩子過篩, 稱取2.5 kg加入每個土缽中備用。
棉花種子(陸地棉Jin 668)用0.1%氯化汞消毒, 在棉花無菌苗培養基中萌發, 煉苗后轉入上述土缽中, 每個土缽種1棵植株, 平行設置不種棉花的土缽土壤, 所有的土壤均用蒸餾水灌溉。植株在溫室條件下[(28±2)℃, 約16 h/8 h光/暗光周期]生長, 在進入六葉期, 對種植棉花的土缽和不種棉花的土缽進行干旱處理(不澆水), 以正常澆水的有棉花的土缽和無棉花的土缽為對照, 干旱處理2周后, 干旱處理的棉花植株發生萎焉, 此時收集各組土壤樣品, 有棉花種植的土壤微生物樣品由根表土壤微生物和根圍土壤微生物組成。棉花根表土壤微生物的收集方法如下: 小心收集干旱和對照棉花的根系, 然后放置在含有25 mL磷酸鹽緩沖液(每升6.33 g NaH2PO4·H2O, 16.5 g Na2HPO4·7H2O, 200 mL Silwet L-77)的50 mL無菌試管中, 高速離心去除根表土壤; 將上層溶液通過100 mm的尼龍膜過濾器過濾以去除殘渣, 然后在5000 ×轉速下離心15 min, 收集下層含有微生物的沉淀, 迅速在液氮中冷凍, 然后–80℃儲存。根圍土壤樣品的收集: 在棉株周圍(無棉株)土壤中6個方向均勻取樣, 15個土缽為一個重復進行混合, 然后收混集10 g土壤樣品放入無菌離心管中。共獲得4種土壤微生物樣品, 分別為干旱處理的棉花根際土壤樣品(drought treated plant rhizosphere soil, SDP)、正常澆水棉花根際土壤樣品(normally watered plant rhizosphere soil, SPN)、未種棉花干旱處理的土壤(drought treated soil without plants, SOPD)和正常澆水的土壤(normally watered soil without plants, SNPN)樣品。每次處理進行3次生物重復, 共收集12個樣品。
1.2 DNA提取、真菌ITS1區擴增、Illumina文庫的制備和高通量測序
由上海派森諾生物科技股份有限公司(中國上海)完成樣品微生物DNA的提取、Illumina文庫的制備和高通量測序。通過引物ITS5F (5¢-GGAAGTAAAAGTCGTAA CAAGG-3¢)和ITS2R (5¢-GCTGCGTTCTTCATCGATGC-3¢)擴增真菌ITS1區, 采用Illumina MiSeq測序平臺進行雙末端(Paired-end)測序。
1.3 原始數據處理、操作分類單位(OTU)劃分及多樣性分析
利用QIIME 1.8.0軟件對測序數據進行識別、拼接, 并去除低質量、非特異擴增序列及嵌合體序列[29]。在相似性97%的水平上使用UCLUST對獲得的序列進行操作分類單元(operational taxonomic unit, OTU)聚類分析, 并篩選出OTUs的代表性序列[30]; 基于UNITE數據庫(Release 5.0, https://unite.ut.ee/)[31], 對OTUs代表序列進行物種注釋分析, 獲得每個OTUs分類學信息并構建稀釋性曲線、物種積累曲線和等級豐度曲線, 利用QIIME軟件計算α多樣性指數, 包括豐富度指數Chao1、多樣性指數Shannon、Simpson指數和ACE指數[32]。物種積累曲線和等級豐度曲線用R語言繪制。
β多樣性分析用于檢驗不同樣本之間群落結構的相似性, 主要通過主成分分析(principal component analysis, PCA)和多維尺度分析(multidimensional scaling, MDS)進行。
1.4 統計分析
觀察評價原始樣本組間差異大小及其統計學顯著性是通過基于排列的PERMANOVA分析來評估。Adonis/ PERMANOVA多元方差分析使用QIIME軟件進行, 并進行999次置換檢測組間差異是否具有統計學顯著性。UniFrac距離組間或組內差異比較分析是根據樣本分組, 分別對加權的UniFrac距離矩陣的組內和組間距離均值進行檢驗, 并通過1000次蒙特卡羅置換檢驗判斷統計顯著性。
2 結果與分析
2.1 高通量測序結果分析
為了探索正常和干旱條件下棉花根際真菌群落, 對正常和干旱條件下的棉花根際土壤和無棉花種植的土壤共12個樣品進行菌群DNA抽提, 利用Illumina高通量測序技術對真菌ITS1區進行測序, 共獲得568,592條Clean tags, 并進行OTUs的劃分。基于≥97%的相似度水平, 通過聚類共獲得970個用于物種分類的OTUs, 其中SNPN、SOPD、SPN和SDP的OTUs總數分別為481、528、743和752個, 其中288個OTUs為所有組共有(圖1-A)。在OTU鑒定后, 對每個樣品的OTUs劃分為門、綱、目、科、屬和種5個分類水平, 結果表明, 相對無棉花種植的土壤, 棉花根際土壤真菌群落更豐富; 另外, 相對于對照, 干旱處理也增加了土壤中微生物的多樣性(圖1-B)。
A: 樣本OTUs數量的Venn圖; B:不同分類級別下樣本OTUs數。SNPN: 正常澆水的土壤樣品; SOPD: 未種棉花干旱處理的土壤樣品; SPN: 正常澆水棉花根際土壤樣品; SDP: 干旱處理的棉花根際土壤樣品。
A: Venn diagram of sample OTUs quantity; B: the number of OTUs of sample at different classification level. SNPN: normally watered soil without plants; SOPD: drought treated soil without plants; SPN: normally watered plant rhizosphere soil; SDP: drought treated plant rhizosphere soil.
2.2 α多樣性分析
通過進行α多樣性分析檢查單個微生物生態系統中物種豐富度和均勻度。真菌群落稀疏曲線結果顯示, 序列量到5000時各樣品稀釋曲線均基本趨于平緩, 表明取樣基本合理, 真實環境中真菌群落結構的置信度較高, 能夠比較真實地反映土壤樣本的真菌群落(圖2-A)。同時在抽取相同序列的條件下, SOP和SPN相對SOPN和SNPN樣本的OUT數多, 說明SOP和SPN樣本中物種豐富度更高。此外, 物種累積曲線也顯示了樣本量足以反映種群的豐富度(圖2-B)。等級豐度曲線也表明, SPN和SDP具有更高的均勻度和豐富度, 而SNPN和SOPD在OTUs上的豐度差異更大且均勻度較低(圖2-C)。
許多指數可以反映微生物群落的α多樣性。Shannon指數反映群落中物種的變化度, Shannon指數高代表群落中物種豐富度高且分布均勻, 常用的測量指標還包括反映群落豐富度的Chao 1指數和ACE指數, 以及反映群落均勻度的Simpson指數。分別計算每個樣本的α多樣性指數, 并在表1中列出。結果顯示, SNP和SDP的α多樣性指數與對照相比均顯著地提高, 表明SPN和SDP具有較高的物種豐富度和均勻度。
2.3 分類學分析
基于OTU分類結果, 獲得了門、綱、目、科、屬和種分類水平下每個樣品的特有組成。不同樣品在每一個分類級別下的微生物類群都有差異, 總體上SDP在綱、目、屬和種水平上比其他樣本具有更高的微生物類群數量(表2)。
A: 稀疏曲線; B: 物種累積曲線; C: 豐度等級曲線。縮寫同圖1。
A: sparse curves, B: species cumulative curve; C: rank abundance curve. Abbreviations are the same as those given in Fig. 1.
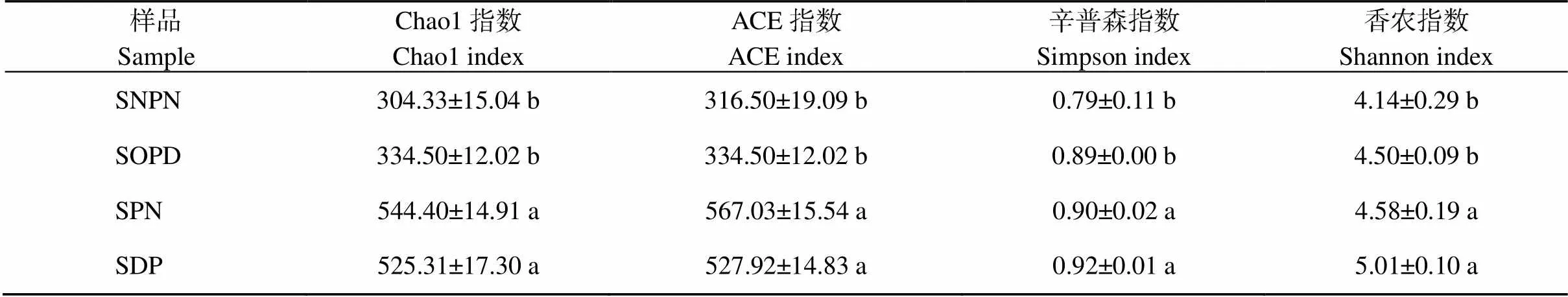
表1 微生物多樣性指數
同列不同小寫字母表示差異顯著(< 0.05)。縮寫同圖1。
Different lowercase letters in the same column indicate significant differences at< 0.05. Abbreviations are the same as those given in Fig. 1.
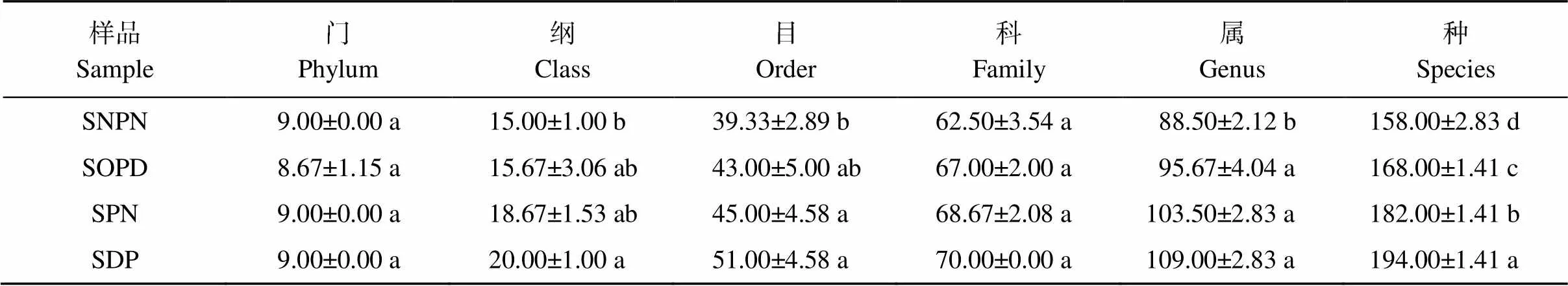
表2 各分類水平的微生物類群數統計表
同列不同小寫字母表示差異顯著(< 0.05)。縮寫同圖1。
Different lowercase letters in the same column indicate significant differences at< 0.05. Abbreviations are the same as those given in Fig. 1.
將OTU的代表序列與微生物參考數據庫進行比對可得到每個OTU對應的物種分類信息, 進而獲得了5個分類層次下(門、綱、目、科和屬)每個樣品的組成和豐度分布。分析棉花土壤樣品真菌多樣性發現, 門分類水平上, 大多數真菌是子囊菌門(Ascomycota)、擔子菌門(Basidiomycota)、類原生動物門(Rozellomycota)、接合菌門(Zygomycota)、纖毛亞門(Ciliophora)、壺菌門(Chytridiomycota)、球囊菌門(Glomeromycota), 其中子囊菌門(Ascomycota)為第一優勢門。與無棉花種植的土壤相比, 棉花根際土壤中壺菌門(Chytridiomycota)和球囊菌門(Glomeromycota)豐度顯著降低(圖3-A)。
在綱分類水平上, 大多數真菌是糞殼菌綱(Sordariomycetes)、散囊菌綱(Eurotiomycetes)、座囊菌綱(Dothideomycetes)、傘菌綱(Agaricomycetes)、圓盤菌綱(Orbiliomycetes)、銀耳綱(Tremellomycetes)。與無棉花種植的土壤相比, 散囊菌綱(Eurotiomycetes)在棉花根際土壤中的豐度顯著增加, 而糞殼菌綱(Sordariomycetes)和座囊菌綱(Dothideomycetes)的豐度顯著降低。此外, 與正常澆水處理的棉花根際土壤相比, 糞殼菌綱(Sordariomycetes)在干旱處理的棉花根際土壤中的豐度顯著降低(圖3-B)。
A~E表示分類單元在門、綱、目、科和屬水平上的百分比。縮寫同圖1。
A–E represent the percentage of taxa at phylum, class, order, family, and genus level, respectively. Abbreviations are the same as those given in Fig. 1.
在目分類水平上, 大多數真菌是糞殼菌目(Sordariales)、肉座菌目(Hypocreales)、小囊菌目(Microascales)、散囊菌目(Eurotiales)、格孢腔菌目(Pleosporales)、圓盤菌目(Orbiliales)。與無棉花種植的土壤相比, 棉花根際土壤中的散囊菌目(Eurotiales)豐度顯著增加, 而肉座菌目(Hypocreales)和格孢腔菌目(Pleosporales)的豐度顯著降低。此外, 與正常澆水處理的棉花根際土壤相比, 散囊菌目(Eurotiales)在干旱處理的棉花根際中豐度顯著增加, 而糞殼菌目(Sordariales)降低(圖3-C)。
毛殼菌科(Chaetomiaceae)、小囊菌科(Microascaceae)、叢赤殼科(Nectriaceae)、發菌科(Trichocomaceae)、毛球殼科(Lasiosphaeriaceae)、圓盤菌科(Orbiliaceae)是較為豐富的科。相對無棉花種植的土壤, 棉花根際土壤中發菌科(Trichocomaceae)和毛球殼科(Lasiosphaeriaceae)的豐度顯著增加, 而毛殼菌科(Chaetomiaceae)的豐度顯著降低。此外, 在干旱處理的棉花根際中發菌科(Trichocomaceae)的豐度顯著增加, 而毛殼菌科(Chaetomiaceae)降低(圖3-D)。
在屬分類水平上, 腐質霉屬()是棉花根際土壤真菌的優勢菌屬, 其在SPN土壤中相對豐度最高, 而在無棉花種植的土壤中相對豐度最低。此外, 毛殼菌屬()、青霉屬()、節叢孢屬()、曲霉屬()、頂孢霉屬()和莖點霉屬()也是不同處理下土壤中的優勢菌群。與無棉花種植的土壤相比, 曲霉屬()和青霉屬()在棉花根際土壤中占優勢, 而頂孢霉屬()和莖點霉屬()的豐度降低。干旱處理增加了曲霉屬()和青霉屬()的豐度(圖3-E)。
系統發育樹分析表明, 子囊菌門(Ascomycota)和擔子菌門(Basidiomycota)是棉花根際土壤中最豐富的門。此外, 最豐富的綱是糞殼菌綱(Sordariomycetes)和傘菌綱(Agaricomycetes) (圖4-A)。根據分類群的豐度分布或樣品之間的相似程度, 對前50個屬在不同樣品中進行聚類分析, 與無棉花種植的土壤相比, 綠僵菌屬()、曲霉屬()、斜蓋傘屬()、青霉屬()、柄孢殼屬()、節擔菌屬()、根霉屬()、足放線病菌屬()和蛇口殼屬()在棉花的根際土壤中相對豐富(圖4-B)。
2.4 β多樣性分析
為了觀察不同樣本之間真菌群落的相似性, 通過主成分分析(PCA)和多維尺度分析對12個樣本進行β多樣性分析。PCA分析發現, 第1主成分(PCA1)和第2主成分(PCA2)分別可以解釋所有變量的74.15%和13.32%, 2個主成分方差累積貢獻率達到87.47%, 說明其能夠表征微生物群落組成的特征, 進一步分析表明, SPN和SDP棉花根際樣品的微生物群落結構相對相似(圖5-A)。經加權UniFrac PCoA分析顯示, 與沒有種植棉花的土壤樣品(SNPN和SOPD)相比, SPN和SDP樣品的真菌群落結構具有很高的相似性, 并且不同(圖5-B)。為更全面地描述組內和組間的菌群結構差異大小, 計算各組之間和組內加權的UniFrac距離均值。結果顯示, SPN-SNPN和SDP-SNPN之間有很大的UniFrac距離, 然而, SDP-SPN之間UniFrac距離較小, 特別是在SPN、SDP和SOPD組內, UniFrac距離非常小(圖5-C)。
A: 顯示樣本群體中從門到屬水平的所有分類群的層次關系的分類層次樹; B: 綜合群體組成的熱水平圖和聚類分析。縮寫同圖1。
A: the classification hierarchy tree shows the hierarchical relationships of all taxa from the phylum to the genus level in the sample population; B: combined heat level map of the community composition with cluster analysis. Abbreviations are the same as those given in Fig. 1.
同時我們也利用偏最小二乘法判別分析(partial least squares discrimination analysis, PLS-DA)對基于物種豐度矩陣和樣本分組數據構建的高通量測序產生的大量微生物群落數據進行判別分析顯示, SPN和SOPD彼此之間更相似(圖5-D)。此外, 對原始樣本組間的差異還進行了方差分析, 加權UniFrac的R統計值為0.7253。此外, 加權后的UniFrac的值和排列數分別為0.001和999。
A: 主成分分析的二維排序; B: 加權UniFrac的PCoA分析; C: 加權UniFrac距離的多組方框圖; D: PLS-判別式分析。縮寫同圖1。
A: two-dimensional ranking of the PCA analysis; B: weighted UniFrac PCoA analysis; C: multiple sets of box plots for the weighted UniFrac distance; D: PLS-discriminant analysis. Abbreviations are the same as those given in Fig. 1.
3 討論
全球環境變化對植物生長產生不利影響, 微生物組作為植物的第2套基因組, 對植物生長及適逆性非常重要,因此微生物群落在增強植物生長方面的潛在功能使得闡明植物微生物群落對環境變化的響應變得非常重要。微生物群落不是一成不變的, 受到環境以及微生物間和植物與微生物間交流的調節[33]。根系分泌物吸引附近土壤環境中的微生物, 改變的根系分泌物擴散到土壤后, 影響了作物根際的土壤環境, 從而致使作物根際微生物數量及種類發生特定變化。例如, 不同植物之間微生物群落組成有顯著差異[20], 環境條件如干旱、鹽度、酸堿度、土壤結構和肥力也影響微生物群落[16,34]。研究表明, 有機肥處理后土壤中細菌和放線菌的群落多樣性與Shannon指數顯著升高, 且氮肥處理后真菌多樣性水平升高[35]; 干旱脅迫的發生也能夠引起棉花根際土壤中細菌的富集且群落多樣性顯著增加[28]。
在本研究中, 通過微生物基因的高通量測序來檢測干旱和正常條件下棉花根際及無棉花種植的連作棉田土壤中的真菌群落。研究結果表明, 不同處理土壤中真菌群落的主要成分存在差異, 其中分類學上不同種類真菌的相對豐度發生變化, 棉花根際真菌群落結構主要由子囊菌門(Ascomycota)、擔子菌門(Basidiomycota)、類原生動物門(Rozellomycota)、接合菌門(Zygomycota)、纖毛亞門(Ciliophora)、壺菌門(Chytridiomycota)、球囊菌門(Glomeromycota)組成, 其中子囊菌門和擔子菌門是植物根際土壤中最豐富的真菌門, 這一結果與前人研究一致[36]。子囊菌門是土壤微生物群落中最豐富的真菌門, 具有降解有機物質的作用, 在有機質含量高的土壤中含量豐富[26,37]。
聚類分析結果表明, 在正常和干旱條件下, 與無棉花種植的土壤相比, 棉花根際土壤中綠僵菌屬()、曲霉屬()、斜蓋傘屬()、青霉屬()、柄孢殼屬()、節擔菌屬()、根霉屬()等相對豐富。其中, 曲霉屬()和青霉屬()在干旱脅迫下棉花根際土壤中豐度較高。這與前人發現鳳仙花的根際真菌群落中青霉屬, 曲霉屬等優勢種在旱季出現的頻度最高這一研究結果一致[38]。曲霉屬是鹽場高濃度鹽水中本土真菌群落的一部分[39], 具有更高的脅迫耐受性, 其中曲霉屬的構巢曲霉()具有耐旱性[40]。此外, 研究發現曲霉屬和青霉屬有較強的滲透壓耐受性, 耐受性最強的是菌種多屬于曲霉屬[41]。因此, 這些對干旱敏感的微生物, 尤其是在干旱條件下植物根際富集的微生物可能有助于植物抵抗干旱和其他非生物脅迫。
近年來研究已報道, 植物在遭受干旱脅迫時, 根際促植物生長細菌作用的植物能更快速產生高量的ABA, 更快地調控基因表達和代謝, 積累脯氨酸和甜菜堿等滲透調節物質, 或增加抗氧化酶和抗氧化物, 提高抗氧化酶活性, 增強植物耐旱的能力[25]。同時, 激素作為植物和微生物之間的中介, 影響植物微生物群落結構的功能[33], 在植物遭受非生物脅迫時也發揮著積極的作用[42]。如根際促植物生長細菌能夠合成刺激植物細胞生長和分裂的植物激素, 從而促使植物抵御環境脅迫[18]。因此, 我們推測曲霉屬()和青霉屬()真菌可能通過刺激植物合成更多的ABA或植物激素的方式, 參與調節植物在抗旱的信號通路, 以提高植物的抗旱性。此外, 它們對植物抗旱性具體作用機制有待進一步的研究。
本研究還發現, 在干旱和正常條件下, 棉花的種植與否對根際真菌群落結構的α多樣性有不同的影響。棉花的種植提升了土壤真菌的物種多樣性和物種豐富度, SPN和SPD樣品具有較高的均勻度和豐富度, 而SNPN和SOPD的OTUs豐度差異較大且均勻度較低。同時, 在干旱脅迫下, SDP有較高的OTUs。這些結果均表明, 植物根系物質的分泌改變了微生物生態環境, 吸引著附近土壤環境中的微生物富集, 使得根際微生物的物種更多樣, 豐富度更高。這些結果與前人發現棉花根際具有更豐富的細菌群落的結果一致[28]; 另一方面, 干旱脅迫可能通過調節根際分泌物的釋放, 刺激了土壤中一些微生物的生長, 在變化的環境下維持植物適應性[43]。此外, β多樣性結果顯示, SPN和SDP之間的微生物群落結構更相似, 與SNPN和SOPD間差異較大, 同樣, SNPN和SOPD彼此之間更相似。結果表明棉花的種植改變了根際土壤微生物群落結構。棉花根際真菌群落結構的變化可能主要受到棉花根與土壤相互作用的影響[28,33,43], 植物根系改變土壤的物理和化學性質, 從而致使根際微生物數量及種類發生特定變化[44]。無棉花種植的土壤(SNPN和SOPD)和根際土壤(SPN和SDP)微生物多樣性的差異分析表明, 微生物附著在根上, 可能隨后促進植物的生長發育、增強耐旱性。此外, 在這項研究中, 鑒定出的許多真菌與棉花植物之間的具體關系是未知的, 因此, 需要進行更多的研究, 尤其是側重于天然存在的根際真菌, 這些真菌是生物防治劑的潛在來源。
[1] Rapparini F, Pe?uelas J. Use of Microbes for the Alleviation of Soil Stresses, New York: Springer, 2014. pp 21–42.
[2] Parida A K, Dagaonkar V S, Phalak M S, Umalkar G V, Aurangabadkar L P. Alterations in photosynthetic pigments, protein and osmotic components in cotton genotypes subjected to short-term drought stress followed by recovery., 2007, 1: 37–48.
[3] Ullah A, Sun H, Yang X Y, Zhang X L. Drought coping strategies in cotton: increased crop per drop., 2017, 15: 271–284.
[4] Rovira A D. Plant root exudates., 1969, 35: 35–57.
[5] Bai Y, Muller D B, Srinivas G, Garrido-Oter R, Potthoff E, Rott M, Dombrowski N, Munch P C, Spaepen S, Remus-Emsermann M, Huttel B, McHardy A C, Vorholt J A, Schulze-Lefert P. Functional overlap of the Arabidopsis leaf and root microbiota., 2015, 528: 364–369.
[6] Wei F, Zhao L, Xu X, Feng H, Shi Y, Deakin G, Feng Z, Zhu H. Cultivar-dependent variation of the cotton rhizosphere and endosphere microbiome under field conditions., 2019, 10: 1659.
[7] Toju H, Peay K G, Yamamichi M, Narisawa K, Hiruma K, Naito K, Fukuda S, Ushio M, Nakaoka S, Onoda Y, Yoshida K, Schlaeppi K, Bai Y, Sugiura R, Ichihashi Y, Minamisawa K, Kiers E T. Core microbiomes for sustainable agroecosystems., 2018, 4: 247–257.
[8] Lau J A, Lennon J T. Rapid responses of soil microorganisms improve plant fitness in novel environments., 2012, 109: 14058–14062.
[9] Niu X, Song L, Xiao Y, Ge W. Drought-tolerant plant growth-promoting rhizobacteria associated with foxtail millet in a semi-arid agroecosystem and their potential in alleviating drought stress., 2017, 8: 2580.
[10] de Zelicourt A, Al-Yousif M, Hirt H. Rhizosphere microbes as essential partners for plant stress tolerance., 2013, 6: 242–245.
[11] Yang J, Kloepper J W, Ryu C M. Rhizosphere bacteria help plants tolerate abiotic stress., 2009, 14: 1–4.
[12] 趙衛松, 郭慶港, 李社增, 王培培, 鹿秀云, 蘇振賀, 張曉云, 馬平. 花鈴期棉花黃萎病抗病與感病品種對土壤細菌群落結構的影響. 中國農業科學, 2020, 53: 942–954.
Zhao W S, Guo Q G, Li S Z, Wang P P, Lu X Y, Su Z H, Zhang X Y, Ma P. Effect of wilt-resistant and wilt-susceptible cotton on soil bacterial community structure at flowering and boll stage., 2020, 53: 942–954 (in Chinese with English abstract).
[13] Edwards J, Johnson C, Santos-Medellín C, Lurie E, Sundaresan V. Structure, variation, and assembly of the root-associated microbiomes of rice., 2015, 112: 911–920.
[14] Walters W A, Jin Z, Youngblut N, Wallace J G, Sutter J, Zhang W, González-Pe?a A, Peiffer J, Koren O, Shi Q, Knight R, Glavina Del Rio T, Tringe S G, Buckler E S, Dangl J L, Ley R E. Large-scale replicated field study of maize rhizosphere identifies heritable microbes., 2018, 115: 7368–7373.
[15] Zolla G, Badri D V, Bakker M G, Manter D K, Vivanco J M. Soil microbiomes vary in their ability to confer drought tolerance to., 2013, 68: 1–9.
[16] Carvalhais L C, Dennis P G, Badri D V, Kidd B N, Vivanco J M, Schenk P M. Linking jasmonic acid signaling, root exudates, and rhizosphere microbiomes., 2015, 28: 1049–1058.
[17] 吳會會, 鄒英寧, 吳強盛. 干旱脅迫下菌根真菌對枳根系形態、內源激素和土壤結構的影響. 中國南方果樹, 2018, 47(3): 14–17.
Wu H H, Zou Y N, Wu J S. Effects of mycorrhizal fungi on root morphology, endogenous hormones and soil structure of trifoliate orange under drought stress., 2018, 47(3): 14–17 (in Chinese with English abstract).
[18] Pieterse C M, Zamioudis C, Berendsen R L, Weller D M, Van Wees S C, Bakker P A. Induced systemic resistance by beneficial microbes., 2014, 52: 347–375.
[19] Hedden P, Thomas S G. Gibberellin biosynthesis and its regulation., 2012, 444: 11–25.
[20] Finkel O M, Castrillo G, Herrera Paredes S, Salas Gonzalez I, Dangl J L. Understanding and exploiting plant beneficial microbes.,2017, 38: 155–163.
[21] Lundberg D S, Lebeis S L, Paredes S H, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Rio T G D. Defining the coreroot microbiome., 2012, 488: 86–90.
[22] 薛英龍, 李春越, 王蓯蓉, 王益, 劉津, 常順, 苗雨, 黨廷輝. 叢枝菌根真菌促進植物攝取土壤磷的作用機制. 水土保持學報, 2019, 33(6): 10–20.
Xue Y L, Li C Y, Wang C R, Wang Y, Liu J, Chang S, Miao Y, Dang T H. Mechanisms of phosphorus uptake from soils by arbuscular mycorrhizal fungi., 2019, 33(6): 10–20 (in Chinese with English abstract).
[23] Rapparini F, Pe?uelas J. Use of Microbes for the Alleviation of Soil Stresses, New York: Springer, 2014. pp 165–174.
[24] Uehlein N, Fileschi K, Eckert M, Bienert G P, Bertl A, Kaldenhoff R. Arbuscular mycorrhizal symbiosis and plant aquaporin expression., 2007, 68: 122–129.
[25] Miller S H, Browne P, Prigent-Combaret C, Combes-Meynet E, Morrissey J P, O’Gara F. Biochemical and genomic comparison of inorganic phosphate solubilization in Pseudomonas species., 2010; 2: 403–411.
[26] Qiao Q, Wang F, Zhang J, Chen Y, Zhang C, Liu G, Zhang H, Ma C, Zhang J. The variation in the rhizosphere microbiome of cotton with soil type, genotype and developmental stage., 2017, 7: 3940.
[27] Xi H, Shen J, Qu Z, Yang D, Liu S, Nie X, Zhu L. Effects of long-term cotton continuous cropping on soil microbiome., 2019, 9: 18297.
[28] Ullah A, Akbar A, Luo Q, Khan A H, Manghwar H, Shaban M, Yang X. Microbiome diversity in cotton rhizosphere under normal and drought conditions., 2019, 77: 429–439.
[29] Caporaso J G, Kuczynski J, Stombaugh J, Bittinger K, Bushman F D, Costello E K, Fierer N, Pe?a A G, Goodrich J K, Gordon J I, Huttley G A, Kelley S T, Knights D, Koenig J E, Ley R E, Lozupone C A, McDonald D, Muegge B D, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh P J, Walters W A, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data., 2010, 7: 335–336.
[30] Edgar R C, Haas B J, Clemente J C, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection., 2011, 27: 2194–2200.
[31] Koljalg U, Nilsson R H, Abarenkov K, Tedersoo L, Taylor A F, Bahram M, Bates S T, Bruns T D, Bengtsson-Palme J, Callaghan T M, Douglas B, Drenkhan T, Eberhardt U, Duenas M, Grebenc T, Griffith G W, Hartmann M, Kirk P M, Kohout P, Larsson E, Lindahl B D, Lucking R, Martin M P, Matheny P B, Nguyen N H, Niskanen T, Oja J, Peay K G, Peintner U, Peterson M, Poldmaa K, Saag L, Saar I, Schussler A, Scott J A, Senes C, Smith M E, Suija A, Taylor D L, Telleria M T, Weiss M, Larsson K H. Towards a unified paradigm for sequence-based identification of fungi., 2013, 22: 5271–5277.
[32] Chao A, Shen T J. Nonparametric prediction in species sampling., 2004, 9: 253–269.
[33] Eichmann R, Richards L, Sch?fer P. Hormones as go-betweens in plant microbiome assembly., 2021, 105: 518–541.
[34] Verbon E H, Liberman L M. Beneficial microbes affect endogenous mechanisms controlling root development., 2016, 21: 218–229.
[35] Yu C, Hu X M, Deng W, Li Y, Xiong C, Ye C H, Han G M, Li X. Changes in soil microbial community structure and functional diversity in the rhizosphere surrounding mulberry subjected to long-term fertilization., 2015, 86: 30–40.
[36] 趙存鵬, 郭寶生, 劉素恩, 王兆曉, 昭耿, 王凱輝, 耿軍義. 糧棉輪作對土壤中養分及真菌多樣性的影響. 華北農學報, 2017, 32(6): 139–146.
Zhao B C, Guo B S, Liu S E, Wang Z X, Zhao G, Wang K H, Geng J Y. Effect of cotton and grain crops rotation on nutrients contents and diversity of fungi in the soil., 2017, 32(6): 139–146 (in Chinese with English abstract).
[37] Rousk J, Brookes P C, B??th E. Fungal and bacterial growth responses to N fertilization and pH in the 150-year ‘Park Grass’ UK grassland experiment., 2011, 76: 89–99.
[38] 劉宇, 韓淑梅, 宋希強, 丁瓊, 王鵬, 趙瑩. 不同海拔下海南鳳仙花可培養根際真菌和細菌群落的季節性變化. 熱帶生物學報, 2018, 9(1): 50–56.
Liu Y, Han S M, Song X Q, Ding Q, Wang P, Zhao Y. Seasonal variation of microbial communities in the rhizosphere of(Balsaminaceae) at different altitudes., 2018, 9(1): 50–56 (in Chinese with English abstract).
[39] Butinar L, Zalar P, Frisvad J C, Gunde-Cimerman N. The genusmembers of indigenous fungal community in hypersaline waters of salterns., 2005, 51: 155–166.
[40] Beever R E, Laracy E P. Osmotic adjustment in the filamentous fungus., 1986, 168: 1358–1365.
[41] Araújo C A S, Ferreira P C, Pupin B, Dias L P, Avalos J, Edwards J, Hallsworth J E, Rangel D E N. Osmotolerance as a determinant of microbial ecology: a study of phylogenetically diverse fungi., 2020, 124: 273–288.
[42] Fahad S, Hussain S, Bano A, Saud S, Hassan S, Shan D, Khan F A, Khan F, Chen Y, Wu C, Tabassum M A, Chun M X, Afzal M, Jan A, Jan M T, Huang J. Potential role of phytohormones and plant growth-promoting rhizobacteria in abiotic stresses: consequences for changing environment., 2015, 22: 4907–4921.
[43] Qiao Q, Zhang J, Ma C, Wang F, Chen Y, Zhang C, Zhang H, Zhang J. Characterization and variation of the rhizosphere fungal community structure of cultivated tetraploid cotton., 2019, 14: e0207903.
[44] Yan N, Marschner P, Cao W, Zuo C, Qin W. Influence of salinity and water content on soil microorganisms., 2015, 3: 316–323.
Fungi diversity analysis of rhizosphere under drought conditions in cotton
YUE Dan-Dan, HAN Bei, Abid Ullah, ZHANG Xian-Long, and YANG Xi-Yan*
National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan 430070, Hubei, China
Plant rhizosphere microorganisms play important roles in plant growth and the adaptability of plants to adverse environmental stresses. In this study, cotton rhizosphere fungal communities were analyzed under drought conditions, aiming to explore the effects of drought stress on the diversity and community structures of cotton rhizosphere fungi, and to provide a theoretical basis for improving cotton water use efficiency by using beneficial microorganisms. Drought stress was applied to upland cotton (cv. Jin 668) at flowering stage (SDP), while the soil without plants was also subjected to drought (SOPD). Simultaneously, the plants (SPN) and pots without plants (SNPN) regularly watered were used as controls. The soil samples were collected, the microbial DNA was isolated, and Illumina Miseq was conducted for a high-throughput sequencing of fungi ITS1 regions to study the diversity of the rhizosphere fungal communities. As a result, a total of 970 OTUs were identified, and the numbers of fungal OTUs in the samples of SNPN, SOPD, SPN, and SDP were 481, 528, 743, and 752, respectively, among which 288 OTUs were shared by all samples. The OTUs were classified to different levels of phyla, class, order, family, and genus of fungi. The rhizosphere fungal community of cotton was predominantly consisted of the phyla Ascomycota (82.70%) and Basidiomycota (10.15%). The abundance of Sordariomycetes, Sordariales, and Chaetomiaceae decreased, while the abundance of Eurotiales, Trichocomaceae,, andincreased significantly under drought stress. Moreover, the diversity of fungal community in the soil with cotton plants significantly higher than that in the soil without cotton plants. Meanwhile, the fungi community structures of SPN and SDP resembling each other and differing greatly from SNPN and SOPD. These results revealed that the cotton rhizosphere had a rich pool of fungal communities, and drought stress had a significant effect on the abundances and diversity of fungi in cotton rhizosphere. This study provided new insights for the researches of improving drought tolerance of cotton in terms of soil microorganisms.
cotton; drought; fungal community diversity; rhizosphere; high-throughput sequencing
10.3724/SP.J.1006.2021.04162
本研究由國家重點研發計劃項目“大田經濟作物優質豐產的生理基礎與調控”(2018YFD1000907)資助。
This study was supported by the National Key Research and Development Program of China “Physiological Basis and Agronomic Management for High-quality and High-yield of Field Cash Crops” (2018YFD1000907).
楊細燕, E-mail: yxy@mail.hzau.edu.cn
E-mail: ddyue@webmail.hzau.edu.cn
2020-07-20;
2021-01-21;
2021-02-25.
URL: https://kns.cnki.net/kcms/detail/11.1809.S.20210225.1448.011.html
猜你喜歡
少兒科學周刊·兒童版(2021年19期)2021-12-10 14:13:40
小學閱讀指南·低年級版(2021年3期)2021-03-19 06:12:40
小天使·二年級語數英綜合(2020年8期)2020-12-23 04:57:40
小天使·一年級語數英綜合(2020年11期)2020-12-16 02:57:22
學苑創造·A版(2020年3期)2020-04-24 09:21:39
小溪流(畫刊)(2017年11期)2018-01-09 19:15:14
少兒科學周刊·兒童版(2017年5期)2017-06-29 22:24:28
少兒科學周刊·兒童版(2017年5期)2017-06-29 16:46:33
紅領巾·萌芽(2017年5期)2017-06-23 10:35:59
爆笑show(2016年7期)2017-02-09 09:36:13
