頸動脈鞘區節細胞神經瘤1例報告并文獻復習
2021-07-15 09:26:50費喜峰沈李奎陳寒春蔣棟毅王之敏
臨床神經外科雜志 2021年3期
關鍵詞:手術
費喜峰,沈李奎,陳寒春,蔣棟毅,王之敏
頸動脈鞘區的腫瘤按解剖部位可歸屬于咽旁間隙腫瘤,此部位腫瘤少見,不到頭頸部腫瘤的0.8%,大多數為良性腫瘤,且多為神經源性和涎源性腫瘤[1-3]。其中神經源性腫瘤主要包括神經鞘瘤及神經纖維瘤;而頸部節細胞神經瘤則是罕見的神經源性腫瘤,在整個頸部良性腫瘤的占比極低。頸部節細胞神經瘤一般來自植物神經系統,尤其是交感神經[4-5]。上海交通大學醫學院附屬蘇州九龍醫院神經外科于2019年7月收治1例成人頸動脈鞘區節細胞神經瘤患者,經手術后病理檢查證實。本研究對該例患者的臨床資料進行回顧分析,并結合相關文獻復習,探討成人頸部節細胞神經瘤的臨床特征、治療方法及術后相關并發癥。
1 臨床資料
患者,女,23歲,因“發現頸部占位1個月”于2019年7月入院。患者既往體健,在體檢時行頸椎MRI檢查發現左側頸動脈鞘區腫瘤,體表無法觸及腫塊,無吞咽苦難,飲水嗆咳及聲音嘶啞等表現。頸椎MRI檢查顯示,左側頸動脈鞘區見類圓形等T1長T2信號影,FS-T2WI呈高信號,大小約為28.5 mm×13.5 mm,其內信號不均勻,DWI呈高信號,ADC呈高信號,增強掃描呈明顯不均勻強化,考慮為神經源性腫瘤(圖1)。顱頸交界區CTA檢查示,左側咽旁頸動脈鞘區見大小約16 mm×12 mm的類圓形低密度影,內部信號欠均勻,增強掃描示不均勻強化,與頸動脈分界尚清楚,血管顯示清晰,局部血管受壓推移;氣管形態好,居中;兩側甲狀腺形態、密度如常。頸部CTA及三維重建示兩側頸內、外動脈充盈良好,管腔未見明顯狹窄及異常擴張。查體:意識清楚,雙側瞳孔等大等圓、直徑3 mm、光反射靈敏,無眼震,雙側鼻唇溝對稱,四肢肌力、肌張力正常,雙側巴彬斯基征陰性,克氏征陰性。2019年7月18日,完善術前準備后在全麻下行頸部腫瘤切除術,患者取仰臥位,常規消毒鋪單;取左側頸部下頜骨下緣橫切口7 cm, 逐層切開, 沿胸鎖乳突肌內側鈍性分離,拉鉤向外側牽開胸鎖乳突肌及頸動脈鞘;向咽旁間隙探查,可見腫瘤組織位于頸動脈鞘內側,頸外動脈受壓外移,腫瘤包膜完整,腫瘤周邊與舌咽、迷走、舌下神經關系密切;用棉片保護重要神經及血管,從神經間的間隙分離腫瘤體表筋膜,腫瘤組織表面光滑,分離出腫瘤上下極,大小約2 cm×2 cm×3 cm,色澤灰;為保證周邊重要神經及血管安全無損傷,在腫瘤組織包膜下予以分塊切除,最后鏡下全切腫瘤;止血紗覆蓋創面,皮下留置引流管一根,逐層縫合肌肉、筋膜和皮膚。手術過程順利,術后患者清醒拔管后返回病房。腫瘤組織術后病理檢查示節細胞神經瘤(圖2)。

A:矢狀位;B:軸位;C:冠狀位,左側頸動脈鞘區腫瘤,邊界清楚,明顯不均勻強化
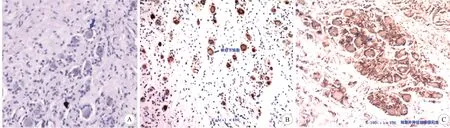
A:腫瘤組織中的節細胞(箭頭所指)(HE染色,×100);B、C:節細胞CGA陽性及腫瘤組織S100陽性表達(箭頭所指)(免疫組化染色,×100)
2 討 論
2.1 頸部節細胞神經瘤及其臨床特征 節細胞神經瘤為少見的良性腫瘤,起源于神經外胚層的神經嵴細胞,占全部周圍腫瘤的2%~3%,多位于后縱隔、腹膜后腔和腎上腺;多為單發病灶,與周圍組織界限清楚,表面一般有完整包膜。節細胞神經瘤大部分無內分泌功能,因此腫瘤在沒有明顯增大之前很少有臨床癥狀;少數存在內分泌功能,腫瘤細胞可分泌如兒茶酚胺、血管活性肽、雄激素等活性物質,而出現高血壓、腹瀉、女性男性化等臨床癥狀[6]。此類腫瘤發生在頸部相當罕見,據已有文獻報道,1980年—1999年19年間僅有5例報道;截至2019年也僅共有21篇文獻報道了27例頸部節細胞神經瘤,且多為兒童患者[7]。頸部節細胞神經瘤主要位于頸動脈鞘區,因處于咽旁間隙,且大多數為良性,極少數會惡變或轉移;生長緩慢,通常無明顯癥狀,病程較長,發現比較困難,經常在體檢時偶然發現;少數因腫瘤較大,壓迫周圍神經和組織后可出現吞咽困難、聲音嘶啞、飲水嗆咳、霍納綜合征等神經損害表現。此處腫瘤多為神經源性和涎源性腫瘤[1-3]。其中神經源性腫瘤主要包括神經鞘瘤及神經纖維瘤。頸部節細胞神經瘤大多來自頸交感干,也有來自迷走神經,可發生于任何年齡,男性與女性發病率基本相同,好發于青少年,大部分發病年齡小于20歲[8-9]。本例患者為成年女性,亦無明顯臨床癥狀,頸部皮膚外觀正常,氣管居中,表面無法觸及包塊,偶然因體檢發現;腫瘤直徑接近3 cm,位于頸動脈鞘區,對頸動脈造成擠壓,屬于節細胞神經瘤在成年患者中的罕見病例。患者在完善術前準備后,行顯微鏡下腫瘤切除術;術后出現了左側Horner綜合征(主要表現為左側眼瞼下垂、瞳孔較健側縮小),無聲音嘶啞及面癱的表現。提示此類腫瘤雖然少見,手術治療方案成熟,但對于術后并發癥的防治,尤其是因腫瘤起源于交感干術后易造成Horner綜合征的防治,臨床醫生仍需高度重視。
2.2 影像及病理學表現 節細胞神經瘤術前診斷困難,如果病灶位于后縱隔、腹膜后或腎上腺,其影像學表現有一定的特征性。如腫瘤常邊界清楚、形態規則、呈偽足或嵌入式生長,CT上表現為低或中等密度,可見鈣化;或MRI見漩渦征,增強掃描呈條索狀或絮狀輕度強化,T2WI顯示高信號等[10-11]。然而,這些特征并非特異,也可見于其他神經源性腫瘤,如神經鞘瘤及神經纖維瘤;因此頸部此類腫瘤常被診斷為神經鞘瘤。同時節細胞神經瘤還應與副交感神經瘤、神經纖維瘤、節細胞神經母細胞瘤相鑒別。最終診斷須依賴病理檢查結果,腫瘤組織含有成熟節細胞是其特征表現。本例患者術前CT檢查示,左側咽旁頸動脈鞘區見16 mm×12 mm大小的類圓形低密度影,內部密度欠均勻,增強掃描呈不均勻強化;MRI檢查示,左側頸動脈鞘區類圓形等T1長T2信號影,其內信號不均,大小約為28.5 mm×13.5 mm,增強掃描呈明顯不均勻強化,考慮神經鞘瘤可能性大。術后病理檢查示腫瘤組織中節細胞成簇分布,胞漿豐富,核大圓形。從病理學角度來說,腫瘤組織中出現節細胞即可診斷為節細胞神經瘤。進一步免疫組化檢查示節細胞表達CGA,神經膜細胞表達S100,而神經鞘瘤和神經纖維瘤一般不表達CGA,最終診斷為節細胞神經瘤。
2.3 手術指征及手術時機的選擇 根據目前文獻報道,主流觀點認為一經發現節細胞神經瘤,無論有無臨床癥狀,均應積極手術切除。如宋揚等[4]報道的6例患者,術前均無臨床癥狀。Xu等[7]報道的7例患者,腫瘤最長徑介于3~15 cm,大多數患者也無臨床癥狀,少數患者有聲音嘶啞等癥狀。本研究認為積極手術治療的理由主要有以下幾點:(1)腫瘤為良性,可達到手術治愈;(2)腫瘤周邊有重要血管及神經豐富,隨著腫瘤的長大,將導致相關功能障礙,并且不容易恢復,甚至無法恢復;(3)腫瘤體積較小時,更容易手術切除,術后并發癥更少。然而,因術后容易出現霍納綜合征,將影響外觀及生活,尤其是在年輕女性患者(如本例患者),故患者本人的手術意愿必須考慮在內,告知患者手術治療的利弊;短期隨訪也是可以選擇的方案。
2.4 手術方式 雖然手術可能存在相應神經損傷,術后出現諸如聲音嘶啞、吞咽困難、面癱、霍納綜合征等并發癥;但因腫瘤為良性,為避免腫瘤生長對周圍重要神經及血管的破壞,手術切除腫瘤仍是目前治療的首選方法[12-16]。手術方式的選擇也種類繁多,主要包括經口入路和經頸入路[17-18];如果腫瘤較大、位置較高,為充分暴露腫瘤則可選擇經頸入路聯合下頜骨掀翻術。經口入路的優勢是神經損傷小,術后無頸部疤痕,美觀;同時在內鏡的配合下使手術更加微創。但是術后患者因無法進食,口腔分泌物增多等因素常有明顯不適感,患者常不愿選擇此方法。經頸入路仍是目前首選的手術方法,同時大部分神經外科醫生對此入路解剖更為熟悉,配合顯微鏡或內鏡可在直視下進行腫瘤切除。為術后美觀,常取頸部橫切口,根據腫瘤的大小及與神經血管的關系,選擇整體切除,或者包膜下分塊切除,盡可能全切腫瘤和保留神經功能,術后頸部切口采取皮內縫合力求美觀。本例患者亦采取經頸入路手術,術中見腫瘤包膜完整,與周邊神經關系密切,從病變位置看,考慮來之椎旁,考慮頸交感干來源;為保護周邊重要神經,采取顯微鏡下包膜下分塊切除,最后在顯微鏡下全切腫瘤。
2.5 術后并發癥 頸部節細胞神經瘤一般多位于上頸部,本例患者腫瘤位于上頸部頸動脈鞘區,此區域包含第9~12對顱神經及頸內外動脈等重要結構,術后容易出現相應神經損傷造成嚴重并發癥。顯微鏡下手術能盡可能地避免重要神經和血管的損傷。然而,由于節細胞神經瘤多來自交感神經,因此手術容易致頸交感神經損傷,霍納綜合征是這類疾病最容易出現的術后并發癥。雖然本例患者是在顯微鏡下包膜下分塊切除腫瘤,手術過程順利;但患者術后仍發生了左側霍納綜合征。霍納綜合征的預后取決于頸交感神經的損傷程度,如果是交感神經間接損傷一般在半年左右能自行恢復,如出現交感神經離斷則無法恢復[4,19-20]。因此,對來源于頸交感干的節細胞神經瘤術后霍納綜合征的預防和治療仍是一個需要進一步探討的問題。本例患者術后早期行高壓氧治療2個療程,輔以針灸及神經營養藥物治療,截至術后3個多月時,患者的眼瞼下垂稍有好轉,遠期恢復仍需進一步觀察。
猜你喜歡
環球時報(2022-12-23)2022-12-23 09:28:37
昆明醫科大學學報(2022年1期)2022-02-28 07:45:04
中老年保健(2021年11期)2021-08-22 03:13:36
昆明醫科大學學報(2021年2期)2021-03-29 07:42:46
河北畫報(2020年10期)2020-11-26 07:20:50
小學閱讀指南·低年級版(2017年1期)2017-03-13 20:07:35
中國衛生標準管理(2015年3期)2016-01-14 03:41:47
中國醫療美容(2015年1期)2015-07-12 10:06:38
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:54
西南軍醫(2014年5期)2014-04-25 07:42:48
