基于HPLC 指紋圖譜結合化學模式識別分析相思子葉質量
2021-06-26 14:10:00何翠敏黃偉斌袁旭江
中成藥 2021年6期
何翠敏,黃偉斌,邱 雨,袁旭江
(廣東藥科大學新藥研發中心/國家中醫藥管理局中藥制劑三級實驗室/廣東省教育廳現代中藥重點實驗室,廣東 廣州 510006)
相思子Abrus precatorius L.為豆科蝶形花亞科相思子屬植物的帶根全草,俗稱印度甘草,原產于印尼,現廣泛分布于世界各地的熱帶和亞熱帶地區,我國廣東、廣西等地均有分布,為習用中草藥,首載于 《千金要方》,其味甘性涼,歸肺、肝、腎經,具有清熱解毒、利尿消炎、潤肺護肝的功效,民間常用于治療咽喉腫痛、肺熱咳嗽、疥瘡乳癰、肝炎等[1-4]。現代研究發現,相思子富含黃酮[5-6],如牡荊素[5]、相思子素[5]、芒柄花黃素[5]、濱薊黃素[6]、濱薊黃苷[6]、異柚葡糖苷[5]等;生物堿[6],如刺桐堿、相思子堿等;三萜[6-8],如甘草酸[6],麥角固醇[7],豆甾醇[7],abrusoside A、B、C、D[8]等;醌[9-10],如abruquinone A、B、D、J、K、L、I、H;氨基酸[11],如賴氨酸、亮氨酸、異亮氨酸等,具有鎮痛抗炎[12]、抗氧化[13]、免疫調節[14]、殺蟲抑菌[15-16]、抗腫瘤[17]等活性。但相思子尚未納入《中國藥典》 中,而相思藤載錄于2004年《廣東省中藥材標準》 第1 版第2 冊,缺乏先進的質控標準,也未見相思子葉指紋圖譜研究,因此,建立先進全面的藥材質量分析和控制方法尤為重要。
指紋圖譜具有系統性、特征性和穩定性,能比較全面地反映中藥所含化學成分的種類和數量,為評價中藥質量、闡明復雜的藥效物質基礎提供科學的技術手段[18]。化學模式識別主要分析數據的差異性,近年來廣泛應用于中藥鑒別、定性表征、質量控制、組效關系等研究中,它對HPLC、UPLCQ-TOF/MS、IR、NMR 等多種現代儀器分析獲取的數據進行客觀分析,既可對多個指標進行統計,又可將整個圖譜信息數量化,被計算機識別與處理,從而可以更加客觀地反映中藥質量信息,達到全面控制評價的目的[19-20]。前期已經開展了相思子葉成分分離、提取工藝、含量測定等相關研究[6,21-23],本實驗將在此基礎上采用HPLC 技術建立藥材指紋圖譜,結合聚類分析、主成分分析評價不同來源藥材質量情況,進一步結合熱圖分析揭示其質量差異與成分的關系,以期為其質量評價和今后開發研究提供參考。
1 材料
Agilent 1100 型高效液相色譜儀(含DAD 檢測器,美國Agilent 公司);JJ500Y 型電子分析天平(0.01 g,廣州市晶博電子有限公司);BS124S 型電子分析天平(0.000 1 g,德國Sartorius 公司);KQ3200 型超聲波清洗器(昆山市超聲儀器有限公司)。濱薊黃苷對照品為廣東藥科大學新藥研發中心自制,純度>99%。甲醇、乙腈為色譜純;甲酸等其余試劑均為分析純;水為超純水。14 批相思子葉采自廣東、廣西和越南,經廣東藥科大學新藥研發中心袁旭江副研究員鑒定為豆科植物相思子Abrus precatorius L.的干燥葉子。具體信息見表1。

表1 樣品信息Tab.1 Information of samples
2 方法與結果
2.1 色譜條件 Agilent ZORBAX SB-C18色譜柱(4.6 mm×250 mm,5 μm);流動相0.1% 甲酸(A)-(甲醇-乙腈)(8∶2)(B),梯度洗脫,程序見表2;體積流量1.0 mL/min;柱溫25 ℃;檢測波長275 nm;進樣量10 μL。

表2 梯度洗脫程序Tab.2 Gradient elution programs
2.2 供試品溶液制備 取干燥相思子葉粉末1 g,精密稱定,置于錐形瓶中,加50%乙醇50 mL,加熱回流提取2 次,每次1 h,合并2 次濾液并濃縮,50%乙醇定容至50 mL 量瓶中,即得(質量濃度為20 mg/mL)。
2.3 對照品溶液制備 取105 ℃烘箱中干燥至恒定質量的濱薊黃苷對照品適量,精密稱定,加50%乙醇溶解并定容至50 mL,即得(質量濃度為0.05 mg/mL)。
2.4 方法學考察
2.4.1 精密度試驗 以濱薊黃苷色譜峰為參照峰(S),取供試品溶液(S1),在“2.1”項色譜條件下連續進樣6 次,計算得到各共有峰相對保留時間RSD 小于0.35%,相對峰面積RSD 小于2.4%;取同一供試品溶液(S1),在“2.1”項色譜條件下連續進樣3 次,重復3 d,測得各共有峰相對保留時間RSD 小于0.49%,相對峰面積RSD 小于3.8%,表明儀器精密度良好。
2.4.2 穩定性試驗 以濱薊黃苷色譜峰為參照峰(S),取供試品溶液(S1),在“2.1”項色譜條件下于0、1、2、4、6、8、12 h 進樣測定,測得各共有峰相對保留時間RSD 小于0.30%,相對峰面積RSD 小于2.7%,表明溶液在12 h 內穩定性良好。
2.4.3 重復性試驗 以濱薊黃苷色譜峰為參照峰(S),取同一批藥材(S1)6 份,按“2.2”項下方法制備供試品溶液,在“2.1”項色譜條件下進樣測定,測得各共有峰相對保留時間RSD 小于0.48%,相對峰面積RSD 小于3.4%,表明該方法重復性良好。
2.5 指紋圖譜建立
2.5.1 圖譜生成 取14 批樣品的供試品溶液,在“2.1”項色譜條件下進樣測定,將數據導入“中藥色譜指紋圖譜相似度評價系統(2004A 版)”,以S12 號色譜圖為參考,中位數法建立對照圖譜,時間窗寬設為0.1 min,采用多點校正Mark 峰匹配,建立指紋圖譜,見圖1。

圖1 14 批樣品色譜圖Fig.1 Chromatograms of fourteen batches of samples
2.5.2 特征峰標定 根據保留時間標定特征指紋峰,對14 批樣品HPLC 指紋圖譜進行分析,發現14 個特征峰為其共有峰,由此確定為特征指紋峰,峰面積總和大于90%。通過對14 個特征峰的紫外光譜圖進行分析歸類,可知除了峰1 和峰2 為生物堿外,其余均為黃酮類成分,峰3~7、10 為二氫黃酮,峰8~9、11~14 為黃酮,進一步查閱文獻和對照品比對,標認峰1 為相思子堿,峰2 為刺桐堿,峰12 為濱薊黃苷,峰14 為濱薊黃素。由于濱薊黃苷(圖1 中12 號)色譜峰的峰面積和峰高適中,峰型對稱,分離度高,故以其為參照峰(S),測得共有峰相對保留時間RSD 為0.14%~0.45%,相對峰面積RSD 為19%~118%,見表3,表明不同來源樣品成分種類穩定,但在含量方面具有差異。

表3 14 批樣品14 個共有峰的相對保留時間、相對峰面積RSDTab.3 Relative retention time and relative peak area RSDs of fourteen common peaks in fourteen batches of samples
2.6 指紋圖譜評價
2.6.1 相似度分析 將14 批樣品指紋圖譜數據導入“中藥色譜指紋圖譜相似度評價系統(2004A版)”,計算相似度,結果見表4,可知S1、S6、S10~S14 相似度最高,均在0.99 以上;S2、S3、S4、S7、S9 樣品次之,介于0.949~0.985 之間;S5、S8 最低,分別為0.879、0.858,表明不同來源樣品的整體質量較為穩定,但個別存在一定差異。

表4 14 批樣品相似度Tab.4 Similarities of fourteen batches of samples
2.6.2 聚類分析 采用SPSS 26.0 軟件進行聚類分析,將14 批樣品HPLC 色譜圖中的14 個共有峰峰面積標準化,形成14×14 原始數據矩陣,采用Ward 聯接方法(離差平方和法),以平方歐氏距離為測度,根據樣品的相似程度進行分類,結果見圖2。由此可知,當判別距離為12 時,14 批樣品分為3 類,S9、S11~14 為第Ⅰ類,除S9 產自越南,其余均產自廣西;S2、S4、S6~S7、S10 為第Ⅱ類,S6~S7 產自廣東,其余均產自廣西;S1、S3、S5、S8 為第Ⅲ類,S3、S8 產自廣東,其余均產自廣西,表明兩廣藥材質量沒有明顯地域分類;產自廣西玉林的樣品(S5、S4、S13、S14)在Ⅰ~Ⅲ類中均有分布,批間穩定性有差異,可能與栽培、采收、加工方式等不同有關。

圖2 14 批樣品聚類樹狀圖Fig.2 Dendrogram of fourteen batches of samples
2.6.3 主成分分析 將14 批樣品14 個共有峰的峰面積進行標準化處理后,導入SPSS 26.0 軟件進行主成分分析,結果見圖3、表5。根據特征值>1提取出4 個主成分(Principal Component,PC),累積貢獻率為85.99%,包含了該指紋圖譜的大部分信息。碎石圖顯示,4 種主成分坡度較陡,是評價藥材質量的標志性成分。由于PC1、PC2、PC3的累積貢獻率達74.24%,可代表藥材指紋圖譜的大部分信息,反映了其基本特征,故以三者得分繪制三維散點圖,見圖4,可見樣品大多數聚在一起,其中S5、S8 和其他樣品的距離相對較遠,與指紋圖譜相似度、聚類分析結果一致。
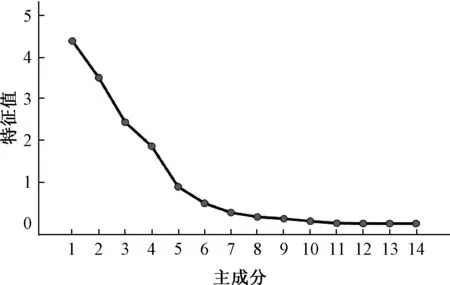
圖3 14 批樣品主成分分析碎石圖Fig.3 Gravel graph of principal component analysis for fourteen batches of samples

表5 主成分初始特征值和方差Tab.5 Initial eigenvalues and variances of principal components

圖4 14 批樣品主成分分析圖Fig.4 Principal component analysis plot for fourteen batches of samples
2.6.4 熱圖分析 由圖5 可知,275 nm 波長下色譜圖最具代表性,各色譜峰分離度均大于1.5,塔板數大于10 000;共有峰5~7、11~12 的峰面積之和占總共有峰峰面積的92.41%;峰12 峰面積顏色變動最大,其次為峰5、6、11、7,其余各色譜峰峰面積較小,顏色變化不明顯,影響程度依次為峰12>5>6>11>7,其余9 個小峰影響程度依次為峰14>13>2>1>9>8>10>3>4。

圖5 14 批樣品共有峰峰面積聚類熱圖Fig.5 Cluster heat maps of the peak areas of fourteen batches of samples
綜上所述,峰5~7、11~12 是藥材最主要成分,其次為1~2、13~14,可作為質量評價指標。
3 討論
本實驗考察了不同型號色譜柱Hypersil BDS C18(4.6 mm×250 mm,5 μm)、Hypersil BDS C18(4.6 mm×250 mm,5 μm)、Agilent ZORBAX SBC18(4.6 mm×250 mm,5 μm)、JADE-PAK ODSAQ(4.6 mm×250 mm,5 μm),有機相甲醇、乙腈、甲醇-乙腈(8∶2),流動相純水、0.1%甲酸、0.3% 甲酸、0.5% 甲酸、1.0% 甲酸,體積流量0.6、0.8、1.0、1.2 mL/min 對色譜峰分離度的影響,并比較了不同檢測波長下色譜峰數量及峰高,最終確定為“2.1”項下條件。
在14 批樣品HPLC 指紋圖譜中,以濱薊黃苷為參照峰(S)確定了14 個共有峰,相似度除了2批相對較低外,其余均在0.949 以上,表明相思子葉整體質量相對穩定。聚類分析、主成分分析結果顯示,樣品質量差異可分成3 類,但與地域無關,關鍵在于各成分含量及其比例是否穩定,而這種穩定可能與采收期有關。熱圖分析結果顯示,樣品質量差異依次與9 種成分色譜峰有關,可作為日后相思子葉質量控制和評價的候選指標。
綜上所述,本研究首次建立了相思子葉HPLC指紋圖譜評價和化學模式識別分析,該方法穩定、快速、可靠,能夠有效、準確地評價相思子葉質量情況及其與成分的關系。
猜你喜歡
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
電子制作(2018年18期)2018-11-14 01:48:24
產品可靠性報告(2017年7期)2017-09-05 09:49:12
山東工業技術(2016年15期)2016-12-01 05:31:22
汽車觀察(2016年3期)2016-02-28 13:16:26
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
中國質量與標準導報(2014年1期)2014-02-28 22:21:28
