乳酸菌基因組代謝網絡模型的研究進展
2021-06-07 02:01:52艾連中侯成杰
食品科學技術學報 2021年3期
關鍵詞:模型
艾連中,侯成杰
(上海理工大學 醫療器械與食品學院/上海食品微生物工程技術研究中心, 上海 200093)
隨著基因組測序技術的快速發展,基因組測序數據量成指數級增長,大量微生物的基因組序列被公布,人們可以從基因和分子水平對微生物的代謝機理進行深入研究,極大地推動了相關學科的發展。如何有效利用這些高通量數據,獲得對生命行為機制的系統理解則成為“后基因組時代”的巨大挑戰[1]。生物系統的動態行為是不同性質的多個分子之間非線性相互作用的結果,而計算機模擬可以高效完成這一工作。基于模擬的研究有助于理解生命系統復雜的底層結構,幫助研究人員對生命行為進行預測。而基因組代謝網絡模型(genome-scale metabolic models,簡稱GEMs或GSMM)正是基于這一需求而發展起來的。基因組代謝網絡模型是基于特定生物體的基因組序列,將所有與代謝相關的基因、酶、生化反應和代謝物數據整合到一起的知識庫,并將其轉化為數學模型,實現對目標生物體代謝表型的預測,系統表征基因與表型之間的關系[2]。
基因組代謝網絡模型有效地模擬了基因組信息和代謝表型之間的關系,為代謝有關的實驗數據提供了堅實的解釋框架,并使全細胞代謝的模擬實驗變得簡單。自1999年世界上第一個基因組代謝網絡模型流感嗜血桿菌模型被構建以來[3],截至2019年已有超過6 000個GEMs被構建[4],并被廣泛應用于系統生物學[5]、代謝工程[6]、藥物開發[7-8]、酶功能預測[9]及微生物群落的相互作用[10]等多個領域。乳酸菌作為一類重要的工業菌,廣泛應用于發酵食品工業,特別是乳制品工業,部分乳酸菌因具有益生功能還被應用于醫學健康領域。為了更深入地對乳酸菌開展功能機制及代謝相關研究,國內外研究人員已構建了多個乳酸菌的GEMs。這些乳酸菌GEMs的構建使人們可以探索乳酸菌在不同環境中的代謝規律和代謝機理,為乳酸菌的研究和應用提供了有價值的工具。
本文將系統回顧微生物基因組代謝網絡模型的構建方法及發展,重點聚焦乳酸菌基因組代謝網絡模型的研究進展,并對未來研究趨勢進行了展望。
1 基因組代謝網絡模型的構建方法
隨著基因測序技術和算法的發展,研究人員能夠基于特定微生物的基因組測序數據和注釋結果構建GEMs。GEMs構建的核心是建立基因- 酶- 代謝反應(GPR)之間的關系。為了完成模型的構建,研究人員需要回答以下問題:酶的底物和產物是什么?是否有多個基因參與同一個酶的表達?每個代謝物的化學計量系數是多少?反應是否可逆?反應在細胞內的定位(細胞質或細胞周質)[11]?這些信息可以通過各種生化數據庫、文獻以及實驗數據來確定。通過建立生化反應組成的網絡,就構成了一個針對特定生物體的代謝網絡模型。構建過程通常包含4個階段,見圖1。
1.1 構建模型草圖
基于基因組注釋構建模型草圖。構建基因組代謝網絡模型的起點是目標生物的基因組注釋信息,這些數據可以通過測序得到,也可以在NCBI等公共數據庫下載。基因組注釋為構建提供了唯一的標識,列出了被認為可能存在于目標生物中的酶,并指出這些基因產物如何相互作用(亞基、復合酶、同工酶)形成具有催化代謝反應的活性酶[11]。代謝數據庫(如KEGG[12]、BRENDA[13]、SEED[14]、Transport DB[15])收集了一系列不同生物體中的代謝反應和轉運反應,為建立酶與代謝反應的關系提供了重要的參考。基于基因組注釋和代謝反應整理所建立的GPR關系就構成了代謝模型的草圖。在構建模型的過程中還需要注意,即使酶的EC號相同,不同生物體之間也會存在底物特異性和酶活性不同,因此,酶在目標生物中催化的反應可能不同于數據庫中的反應。
1.2 人工精煉
代謝模型草圖的人工精煉。代謝模型草圖雖然提供了基于基因注釋的候選反應合集,但還不能建立目標生物體特有的特征。這些特征信息需要根據目標生物體的文獻信息進行人工校正。同時,模型草圖中還包含了一些錯誤反應和代謝缺口(gap),因此必須通過人工精煉以修正錯誤反應,填補缺失反應。模型中所有反應的化學計量學平衡、電荷平衡、元素平衡都需要進行人工檢驗。因此人工精煉是整個GEMs構建過程中最耗時耗力的,甚至是煩瑣的步驟。代謝網絡模型的精煉通常需要數月到一年的時間才能完成,這既取決于目標生物的基因組大小,也取決于目標生物是否有足夠數量的生化數據[16]。文獻、教材、目標生物特有的數據庫以及熟悉目標生物的專家都是人工精煉步驟的重要信息來源。
一個高質量的GEMs是通過基因組注釋和人工管理相結合的方式構建的。這一過程將創建一個針對目標生物的生化、基因組、遺傳學和表型的知識庫。而隨著新的基因注釋結果和新的實驗數據的發表,目標生物的代謝網絡模型應以迭代的方式進行更新[17]。
1.3 模型轉換
將代謝網絡模型轉換為數學模型。精煉后的代謝模型需要轉換為計算機可以識別的數學格式(S矩陣,如圖2),以便通過計算機對模型進行模擬計算。在S矩陣中,行代表網絡代謝產物,列代表代謝反應。反應中的底物被定義為具有負系數,而產物具有正系數[16]。
在進行模型模擬計算前,需要定義模型的系統邊界,也就是對所有能被目標生物消耗或分泌的代謝物,都需要在模型中加入交換反應。交換反應可以在模擬中用于定義環境條件(如碳源、氧氣等)。擬穩態假設和基于約束的重構分析(constraint-based reconstruction and analysis,COBRA)[18]是最廣泛應用的模擬計算策略。對模型進行優化計算最常用的目標函數是優化生長速率,即生物量函數,由生長所需的基本代謝產物組成。利用數學表達式和計算平臺生成生物質目標函數是驗證代謝網絡模型最基本的功能。細胞生物質組成數據可以通過實驗獲得,也可以通過文獻或數據庫獲得。
流量平衡分析(flux balance analysis,FBA)是表征代謝網絡模型最常用的方法。FBA是一種分析代謝網絡中代謝物流動的數學方法,通過計算代謝網絡的代謝物流量,從而可以預測生物體的生長速度或重要代謝物的生成速率[19]。S矩陣和目標函數定義了一個線性方程組,在給定的約束條件下可以求解得到一個解空間。FBA可以識別解空間中優化目標函數的單個最優通量分布或多個最優通量分布。目前,廣泛用于流量平衡分析的軟件是基于Matlab平臺的COBRA工具箱[20]和基于python平臺的COBRApy[21]。
1.4 驗證評估
模型的驗證與評估。精煉后的模型是否能夠準確預測微生物的生長表型是需要進行驗證和評估的。驗證和評估模型的方法有很多,最常用的方法是單一碳源驗證、氨基酸缺失驗證以及基于文獻的各種生理代謝參數驗證[22]。例如:通過限制碳源、氨基酸、生長因子的通量,我們可以預測菌株的營養缺陷型;通過恒化培養實驗與模擬生長速率進行擬合,可以推測出模型中存在的影響生物質合成的代謝途徑[11,16]。將這些模擬計算的結果與“濕”實驗數據進行對比驗證,可以幫助我們分析模型中的缺陷并改進。總之,代謝網絡模型的構建是一個不斷迭代的過程,而如何確定模型構建完成則取決于模型構建的范圍和用途[16]。
為了確保構建結果的準確性和可用性,Palsson課題組于2010年發表了構建代謝網絡模型的標準化操作程序,該程序共分為5個階段,包含了96個具體步驟[16]。該程序很好地規范了構建GEMs的流程及GEMs的格式,使構建GEMs具有了統一的標準,方便國際間的交流與合作。
人工構建基因組代謝網絡模型費時費力,因此多個研究團隊還開發了用于GEMs構建的自動化工具,如ModelSEED[23]、RAVEN[24]、Merlin[25]、CarveMe[26]、kbase[27]等。這些自動化工具大大提高了構建GEMs 的效率,有的工具只需要幾十個小時就可以構建一個GEMs。利用自動化工具構建的GEMs普遍存在大量的錯誤反應、代謝缺口,甚至是基因注釋錯誤,所以這些自動構建的GEMs通常只能作為代謝模型的草圖使用,還需要人工精煉才能成為高質量的GEMs。現有的自動化構建工具尚無法完全替代人工精煉的步驟,目前最常用的方法是自動化構建與人工精煉相結合的方式,即先通過自動化工具構建出目標生物的代謝網絡模型草圖,再由人工對草圖進行精煉和驗證,最終得到一個高質量的GEMs[28-29]。
2 微生物基因組代謝網絡模型的發展歷程
基因組測序數據的指數級增長加速了微生物GEMs的構建,截至2019年2月,已有6 105個微生物基因組代謝網絡模型被構建,其中細菌5 897個,古細菌127個,真核微生物81個[4]。這些快速增加的GEMs中大部分是由自動化工具構建的未人工精煉模型,雖然也被用于各種微生物的研究,但相較于經過人工精煉的高質量GEMs,它們的預測準確度比較差。以大腸桿菌為代表的模式生物,因被科學家廣泛研究而積累了大量生理生化數據,使得這些模式生物在構建GEMs的過程中具有極大的優勢,產生了一系列高質量的GEMs。以大腸桿菌為例,從2000年發布的第一個模型開始到2017年發表的iML1515[30],已經歷了至少6次迭代,模型的規模和準確度都有了極大提升。這些模式菌株GEMs的構建和發展使代謝網絡模型的應用不斷擴大,為目標生物體的代謝研究提供了極好的知識庫,同時也為其它微生物GEMs的構建提供了良好的參考模板,推動更多模型的出現,也推動了構建方法的不斷升級。
2.1 大腸桿菌基因組代謝網絡模型
第一個大腸桿菌(Escherichiacoli)基因組代謝網絡模型是基于大腸桿菌E.coliK-12 MG1655構建的iJE660[31],該模型包含了660個基因,含有完整的氨基酸、核酸、細胞壁和輔因子合成代謝、脂肪酸代謝,能夠對生長表型進行預測,基因缺失模擬的準確度為86%。在后續的幾次迭代中,陸續添加了不同碳源代謝、醌的表征[32]、細胞周質交換反應、完善了元素平衡和電荷平衡,并填充了缺失反應[33],不斷提高模型的準確性,見圖3。最新的iML1515模型包含了1 515個基因,2 719個代謝反應和1 192個代謝物,并加入了所有酶的三維結構。iML1515對基因重要性的模擬準確率達到了93.4%。此外,iML1515還可以從大量生物數據中提取最相關的信息快速生成新的子模型。例如:通過iML1515生成的子集iML976僅包含了1 000多個大腸桿菌菌株共有的代謝網絡信息,使人們可以更清楚的了解大腸桿菌的核心和輔助代謝能力,突出了識別藥物靶標的潛力。將iML1515用于分析不同條件下的轉錄組數據為轉錄組變異分析提供了有價值的見解。由大腸桿菌GEMs的發展可以看出,隨著模型的不斷迭代,其覆蓋的基因和代謝反應數量不斷增加,模型的應用范圍也不斷擴大。早期的GEMs主要用于計算微生物的生長速率、副產物產量等基本表型,而最新的GEMs已經可以用于各種應激反應分析、泛基因組分析、蛋白質組功能分析等[30]。
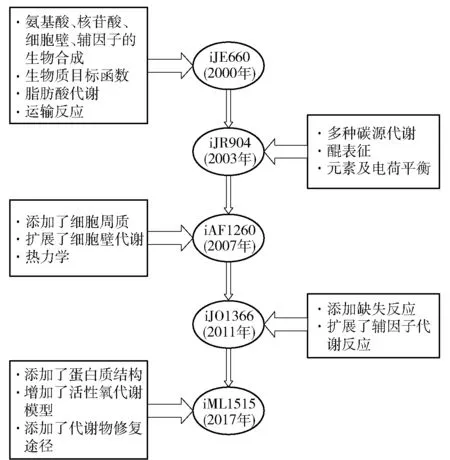
圖3 大腸桿菌GEMs的迭代Fig.3 Iteration of E. coli GEMs
2.2 枯草芽孢桿菌基因組代謝網絡模型
枯草芽孢桿菌(Bacillussubtilis)是革蘭陽性菌中的代表性菌種,具有出色的生產異種蛋白的能力,被認為是工業酶和生物制藥的“細胞工廠”[34]。目前已有多個枯草芽孢桿菌的GEMs被構建,包括iYO844[35]、iBsu1103[36]、iBsu1103V2[37]、iBsu1147[38]、iBsu1144[39]以及天津大學研究團隊構建的iBsu1141[40]。最新版本的枯草芽孢桿菌GEMs是iBsu1144,該模型基于對枯草芽孢桿菌基因組重新注釋而建立。iBsu1144包含了1 144個基因、1 955個反應和1 103個代謝物,通過熱力學分析重新修正了代謝反應的方向性和可逆性。基于必需基因和非必需基因分析的準確率分別達到91.6%和91.4%。該模型可用于枯草芽孢桿菌的代謝工程設計,同時也為其他革蘭陽性菌GEMs的構建提供了參考模板。
2.3 釀酒酵母基因組代謝網絡模型
釀酒酵母(Saccharomycescerevisiae)是第一個被構建GEMs的真核微生物[41]。與原核微生物不同,真核微生物中存在多個細胞器,在構建GEMs時需要考慮代謝反應發生的區室并加入胞內轉運反應,因此真核微生物GEMs的構建過程更加復雜。
釀酒酵母作為真核生物的模式生物而受到廣泛的關注和研究,從釀酒酵母的第一個GEMs[41]誕生以來,已經有多個研究團隊發布了多個版本的釀酒酵母GEMs[42-44]。由于不同研究團隊在注釋結果和術語等方面存在差異,使不同模型之間的比較和使用都變得困難,給釀酒酵母GEMs的應用和升級帶來了極大困擾。為解決這一問題,科學界通過國際合作建立了一個關于酵母的共識代謝網絡模型[45],這一共識模型采用統一的規范術語并由專人維護升級。最新發表的釀酒酵母代謝模型有效整合了RNA和蛋白質合成數據,得到了第一個添加了表達與熱力學通量(ETFL)的釀酒酵母代謝與表達模型(ME-models)。在預測最大生長速率、必需基因和溢出代謝表型等方面表現出良好的能力[46]。
3 乳酸菌基因組代謝網絡模型的構建現狀
乳酸菌是一類能夠將碳水化合物轉化為乳酸的細菌的統稱[47]。乳酸菌在發酵的過程中利用碳源產生乳酸使食品快速酸化,可以延長食品的貨架期和保質期,同時乳酸菌代謝產生的一些化合物還能夠賦予食品特殊的風味和質地[48]。近二十年來,隨著微生物組技術、組學技術和生物信息學技術在乳酸菌領域的應用,人們對乳酸菌的代謝有了深入的認知,構建乳酸菌的GEMs也有助于更深入的研究乳酸菌的代謝活動,指導乳酸菌的代謝工程改造。表1列出了已發表的乳酸菌GEMs[28-29,49-55]。
3.1 乳酸乳球菌基因組代謝網絡模型
乳酸乳球菌(Lactococcuslactis)作為乳酸菌的模式菌株,是研究最廣泛的一種乳酸菌,其基因組也是乳酸菌中第一個被測序和注釋的[56]。第一個乳酸乳球菌GEMs是由Nielsen課題組于2005年構建[49]。該模型是基于Lactococcuslactisssp.lactisIL1403的基因組注釋構建的,包含358個基因、621個反應和509個代謝物,其中476個代謝反應與基因建立關聯,其余145個代謝反應是基于生理生化考慮而推測加入的。通過FBA和MOMA進行代謝分析,證明該模型的很多預測結果和實驗結果是吻合的。例如:在所有氨基酸都被提供的情況下,乳酸乳球菌更傾向于從頭合成丙氨酸、天冬氨酸、甘氨酸和苯丙氨酸。該模型作為一個有用的工具可用于測試或開發新的代謝工程策略[49]。
另一個乳酸乳球菌GEMs是由荷蘭瓦赫寧根大學的研究團隊基于Lactococcuslactisssp.cremorisMG1363構建的,得益于十幾年來分子生物學和基因組學的快速發展,該模型涵蓋的基因數量明顯增加,包括518個基因、754個反應和650個代謝物。通過FBA和FVA分析了整個代謝網絡中的通量分布情況以及與風味物質形成有關的通路,發現754個代謝反應中有59個反應直接或間接參與了風味的形成。全面的模型驅動分析顯示了乳酸乳球菌高度靈活的氮代謝,與氧化還原平衡相結合的支鏈氨基酸分解代謝是預測不同風味化合物形成的關鍵。該模型為解析乳酸菌合成風味物質的代謝網絡提供了基礎工具[50]。
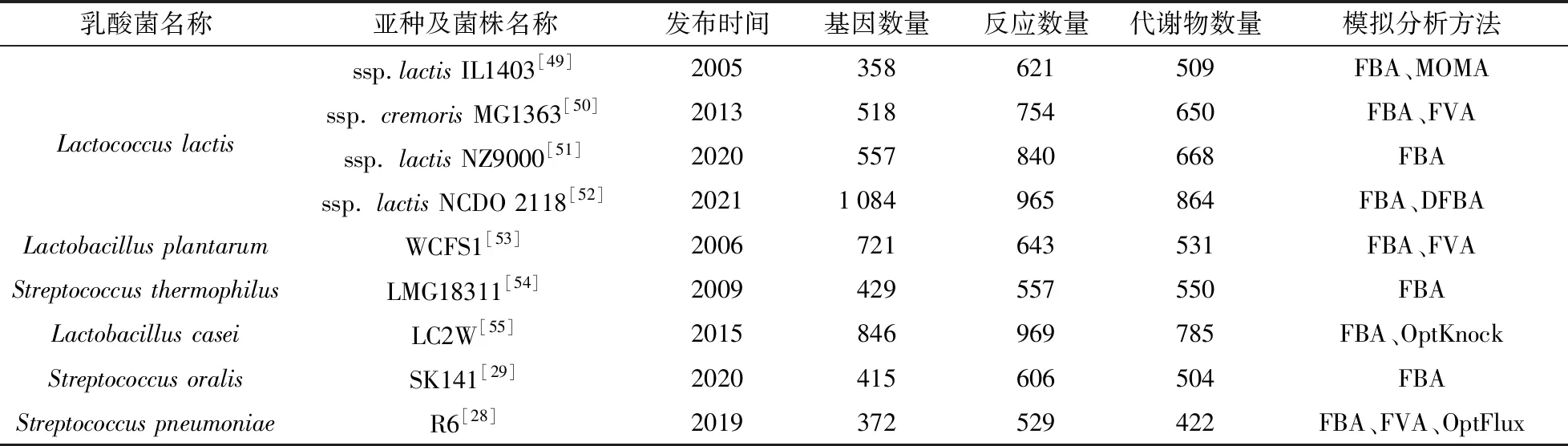
表1 已發表的乳酸菌GEMsTab.1 Published lactic acid bacteria GEMs
最新發表的乳酸乳球菌GEMs(iOA1084)是基于Lactococcuslactisssp.lactisNCDO 2118構建的,該菌株具有高產γ-氨基丁酸(GABA)的能力。iOA1084涵蓋了62個代謝途徑、1 084個基因、965個代謝反應和864個代謝物。研究者重點關注了通過模型預測提高菌株產GABA的能力,通過FBA分析正常和高谷氨酸攝取速率下GABA的產生,發現高谷氨酸可以增加GABA的生產速率[52]。
3.2 植物乳桿菌基因組代謝網絡模型
植物乳桿菌(Lactobacillusplantarum)是一種工業菌株,因其具有益生作用而受到廣泛關注。研究人員基于植物乳桿菌WCFS1構建了一個包含721個基因、643個反應和531個代謝物的GSMs,優化了ATP的產生并確定了與自由能代謝無關的氨基酸分解代謝途徑。通過FVA分析補充了基本模型中的平行途徑,如產物相同但輔因子不同的途徑。FBA分析顯示模型過高估計了增加葡萄糖濃度產生的生物質通量而低估了乳酸生成量,這可能是因為模型假定細胞生成生物質的效率最優,所以不能有效預測代謝效率較低的乳酸的生成[53]。
3.3 嗜熱鏈球菌基因組代謝網絡模型
嗜熱鏈球菌(Streptococcusthermophilus)作為優良發酵劑被廣泛用于乳制品工業,基于嗜熱鏈球菌LMG18311的GEMs于2009年發布并與其他兩種乳酸菌(乳酸乳球菌和植物乳桿菌)的GEMs進行了比較。相較于其它兩種乳酸菌,嗜熱鏈球菌顯示出較低的自養能力,這種低自養能力是由于嗜熱鏈球菌的進化環境決定的。此外,風味分析發現嗜熱鏈球菌氨基酸代謝通路產生了較多的揮發性化合物,這也是其賦予酸奶等發酵乳制品特殊風味的原因[54]。
3.4 干酪乳桿菌基因組代謝網絡模型
干酪乳桿菌(Lactobacilluscasei)被認為是一種具有益生作用的乳酸菌,在乳制品、制藥及臨床醫學領域均有應用。江南大學劉立明團隊利用人工注釋和ModelSeed自動建模結合的方式構建了第一個干酪乳桿菌基因組代謝網絡模型iJL846,該模型由846個基因、969個代謝反應和785個代謝產物組成。模型分析發現有10種氨基酸和7種維生素是干酪乳桿菌LC2W的必需營養素;通量分析表明EMP途徑是產生乳酸的主要途徑,同時預測了混合酸發酵過程中氨基酸的通量。FBA分析表明揮發性風味化合物的形成與氧環境有直接關系,并通過模擬基因缺失預測了三個可以改善3-羥基丁酮產量的新靶點[55]。
3.5 口腔鏈球菌基因組代謝網絡模型
口腔鏈球菌(Streptococcusoralis)是一種存在于口腔中的乳酸菌,與口腔中其他細菌存在著復雜的互作關系,這些互作關系被認為與人類的口腔健康有關。Palsson課題組基于口腔鏈球菌SK141的基因組序列,并結合近緣物種的實驗數據構建了首個口腔鏈球菌的GEMs(iCJ415)。利用近緣物種的基因必需性數據和氨基酸營養缺陷數據驗證該模型,其基因必需性的預測準確率為71%~76%,氨基酸營養缺陷預測準確率為85%,碳源預測結果的準確率為82%。說明iCJ415可以較好地反映口腔鏈球菌的代謝特性,該模型可以作為探索同一物種不同菌株之間相互作用以及不同物種在人類口腔內復雜代謝作用的起點[29]。
3.6 肺炎鏈球菌基因組代謝網絡模型
肺炎鏈球菌(Streptococcuspneumoniae)是一種革蘭氏陽性菌,可以產生少量乳酸,因此也屬于乳酸菌[47]。但不同于大多數乳酸菌,肺炎鏈球菌是一種致病菌,可導致肺炎、中耳炎等疾病。為了更好地了解肺炎鏈球菌的代謝,研究人員利用自動化構建軟件Merlin構建了肺炎鏈球菌的基因組代謝模型iDS372,該模型可以模擬肺炎鏈球菌在不同環境條件下的代謝行為,為新的藥物靶點提供了線索[28]。
4 乳酸菌基因組代謝網絡模型的研究展望
雖然研究人員很早就開始了乳酸菌GEMs的構建,但相較于以大腸桿菌和釀酒酵母為代表的模式菌株,乳酸菌GEMs無論在數量還是質量上都明顯落后。很多模型是十幾年前構建的,模型中往往只包含了GPR關系等核心內容,缺少代謝通路子系統分類,代謝物也沒有進行統一術語規范,不利于其他研究人員對模型進行應用和升級。乳酸菌GEMs的研究還有很大的空間,同時也存在巨大的挑戰。未來乳酸菌GEMs的研究可以重點圍繞以下兩個方面開展。
4.1 更新乳酸菌GEMs,構建乳酸菌ME模型
隨著基因組學、微生物組學的快速發展以及大量乳酸菌生理生化數據的更新,基因注釋的范圍及準確性都有了明顯提高,而現有的乳酸菌GEMs構建時間普遍較早,無論是基因注釋信息、數量以及代謝反應的數量和質量都需要進行更新。隨著蛋白組、轉錄組、代謝組等組學技術的發展,整合代謝模型和蛋白質合成途徑(包括轉錄、翻譯)的ME模型(代謝與大分子表達模型)應運而生。ME模型除了反應代謝網絡外,還明確包含了構成轉錄和翻譯的途徑,能夠模擬蛋白質組的組成,因此,ME模型可以用于計算菌株生長條件或新環境進化適應性的蛋白質組分配,大大擴展了模型的功能和應用范圍。ME模型是代謝模型(M)和表達矩陣(E)的整合,表達矩陣中包含了已知的所有功能成分(蛋白質、核苷酸等)和轉錄途徑,包括RNA和蛋白質的合成、修飾和降解[17]。ME模型不僅可以預測細胞的最大生長速率和相應的代謝通量,還可以計算最優的蛋白質組分配和基因產物表達水平。通過ME模型可以實現組學數據的定量集成和表型模擬。目前已經發表了多個大腸桿菌ME模型[9, 30],但是還沒有針對乳酸菌的ME模型。構建乳酸菌ME模型可以為乳酸菌的代謝工程設計提供更有價值的策略。
4.2 構建乳酸菌泛基因組模型
隨著基因組測序數量的不斷增加,泛基因組的概念被提出。泛基因組是指某一物種全部基因的總和,由核心基因組(即一個物種內所有菌株共有的基因)和輔助基因組(即只存在于某個菌株的基因)構成[57]。泛基因組可用于不同菌株之間的基因比較分析,有助于對菌株代謝過程的所有機制進行更深入的分析。通過對炎癥性腸病患者臨床分離得到的大腸桿菌進行泛基因組分析發現,患者體內分離到的特定大腸桿菌菌株能通過特殊代謝途徑參與黏液多糖代謝,有助于該菌株在腸道內的定植[58]。乳酸菌在腸道中的定植能力與其功能作用有著密切的關系,大腸桿菌的泛基因組分析為乳酸菌在腸道內的定植研究提供了新思路。目前已構建的泛基因組代謝網絡模型主要集中于大腸桿菌、沙門氏菌[59]、金黃色葡萄球菌[60]等致病菌。而乳酸菌中包含數量眾多的益生菌,其在維持人體腸道健康等方面具有重要的作用,建立乳酸菌泛基因組模型將有助于我們解析這些菌株的特異性,研究其在人體腸道定植以及與其他腸道微生物互作的機理。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
