川芎揮發油羥丙基-β-環糊精包合物的制備及其性能研究
2021-06-07 02:47:04楊玉婷劉云華劉玉紅黃志芳易進海
天然產物研究與開發 2021年5期
關鍵詞:方法
楊玉婷,劉云華,劉玉紅,黃志芳,陳 燕,易進海*
1成都中醫藥大學藥學院,成都 611137;2四川省中醫藥科學院 中藥材品質及創新中藥研究四川省重點實驗室,成都 610041
川芎為傘形科藁本屬植物LigusticumchuanxiongHort. 的干燥根莖,其味辛、微苦、性溫,具有活血行氣、祛風止痛等功效,被譽為“血中之氣藥”,用于胸痹心痛,跌撲腫痛,月經不調,癥瘕腹痛,風濕痹痛等癥[1]。川芎揮發油(川芎油)是川芎的重要有效組分之一,主要有效成分為藁本內酯,具有很強的解痙、平喘、鎮靜等作用[2],臨床上治療偏頭痛和缺血性腦血管疾病等有效[3,4]。但藁本內酯的化學性質不穩定,易發生脫氫、氧化、水解、降解等多種異構化反應[5],此外,川芎油水溶性低及口服生物利用度較差,極大地限制了其在臨床上的應用。為解決川芎油穩定性及溶解度的問題,已報道的制劑方法有:軟膠囊、滴丸、鼻腔噴霧劑、微乳、環糊精包合等[3,6]。環糊精包合是一種常用的制劑手段,羥丙基-β-環糊精(hydroxypropyl-β-cyclodextrin,HP-β-CD)是應用最廣泛的環糊精衍生物之一,也是FDA第一個批準可供注射的β-環糊精(β-cyclodextrin,β-CD)衍生物[7]。HP-β-CD是由β-CD與環氧丙烷縮合得到的親水性衍生物,由于β-CD的羥丙基化破壞了其分子內氫鍵,HP-β-CD水溶性顯著提高(75% W/W),同時又保留了β-CD的包合能力,極大地擴寬了母體β-CD的應用范圍,具有提高藥物的溶解度、穩定性和生物利用度,降低藥物的刺激性或毒性等作用[8]。
目前中藥揮發油的包合材料大多選用β-CD,一般采用飽和水溶液法制備包合物[6,9],但收率較低且水溶性差。采用高水溶性的HP-β-CD制備包合物,收率高且水溶性好,常規制備方法有水溶液法、研磨法和超聲法,該3種方法均在水相中進行,需攪拌、研磨或超聲處理,效率低且不易干燥[10-12]。Ao等[13]以無水乙醇為溶劑,采用旋轉蒸發器制備川芎油HP-β-CD包合物,用旋轉代替傳統的攪拌工序,但無水乙醇和旋轉工序難以應用于工業化大生產。其次,該研究采用乙酸乙酯回流提取川芎油,以得到的川芎油重量計算包合率,需要的包合物量大,且提取不完全,精準度差。此外,該研究未對包合物進行表征、溶解度及穩定性評價。本實驗基于大生產的可行性和實用性,擬以85~95%乙醇為溶劑,采用單相法制備川芎油HP-β-CD包合物,無需攪拌、研磨等工序,直接回收濃縮干燥,該方法簡便易操作,且收率和包合率高;研究確定了包合物提取和洗脫方法,采用HPLC準確測定包合率,正交試驗法優化包合工藝,對包合物進行表征、溶解度和穩定性考察,為川芎油制劑的生產應用提供參考。
1 儀器與材料
Agilent 1260高效液相色譜儀(美國Agilent公司);KQ-300E超聲波清洗器(昆山市超聲儀器有限公司);R-3旋轉蒸發儀(瑞士BUCHI公司);Apreo S型掃描電鏡(美國賽默飛世爾科技公司);Cary 600 FTIR光譜儀(美國Agilent公司);ZKF035型電熱真空干燥箱(上海實驗儀器廠有限公司);SOP型電子天平(賽多利斯科學儀器(北京)有限公司);Milli-Q Integral 3型超純水機(美國Millipore 公司);DNP-9052型電熱恒溫培養箱(上海精宏實驗設備有限公司);IKA C-MAG HS7型加熱磁力攪拌器(德國IKA公司);SoRVALL ST16R高速離心機(美國賽默飛世爾科技公司);ZD-85型氣浴恒溫振蕩器(常州國宇儀器制造有限公司)。
川芎油(實驗室自制,藁本內酯、洋川芎內酯A的含量分別為50.30%和16.77%);HP-β-CD(批號:HP20200313,山東濱州智源生物科技有限公司);對照品丁苯酞(批號:101035-201903,中國食品藥品檢定研究院);無水乙醇(分析純,成都市科龍化工試劑廠);甲醇(分析純,成都市科龍化工試劑廠);石油醚(沸程:60~90 ℃,分析純,成都市科龍化工試劑廠);甲醇(色譜純,賽默飛世爾科技(中國)有限公司);水(超純水,Milli-Q Integral 3超純水機制得)。
2 方法與結果
2.1 包合物的制備
2.1.1 HP-β-CD包合物的制備
稱取HP-β-CD適量,溶于適量95%乙醇中,按規定比例加入川芎油,混合均勻后,70 ℃減壓回收乙醇、干燥。研磨、過80目篩,得淡黃色包合物粉末。
2.1.2 HP-β-CD物理混合物的制備
稱取HP-β-CD適量于研缽中,緩慢加入適量川芎油,輕輕研勻,得淡黃色物理混合物粉末。
2.2 含量測定
采用本課題組建立報道的方法[14],以丁苯酞為對照品,一測多評法測定川芎油中藁本內酯和洋川芎內酯A的含量,藁本內酯、洋川芎內酯A對丁苯酞的校正因子分別為0.226 3、0.490 7。
2.2.1 色譜條件[14]
采用Agilent Eclipse XDB-C18色譜柱(4.6 mm×150 mm,5 μm);流動相為甲醇-水(52∶48);柱溫35 ℃;檢測波長280 nm;流速1.0 mL/min;進樣量5 μL。色譜圖見圖1。
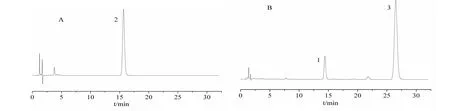
圖1 對照品溶液(A)、川芎油HP-β-CD包合物(B)HPLC色譜圖Fig.1 HPLC chromatograms of reference substance (A) and Chuanxiong Rhizoma volatile oil-HP-β-CD inclusion complex (B)注:1.洋川芎內酯A;2.丁苯酞;3.藁本內酯。Note: 1.Senkyunolide A;2.Butylphthalide;3.Ligustilide.
2.2.2 對照品溶液的制備
取丁苯酞對照品適量,精密稱定,加甲醇制成每1 mL含204.8 μg的溶液,即得。
2.2.3 供試品溶液的制備
取包合物粉末約0.1 g,精密稱定,置具塞錐形瓶中,精密加入70%乙醇25 mL,密塞,稱定重量,超聲處理(功率300 W,頻率40 kHz)30 min,放冷,再稱定重量,用70%乙醇補足減失的重量,搖勻,濾過,取續濾液,即得。
2.3 包合率的測定
包合率為衡量包合效果的主要指標,包合率越高,揮發油被包合的效果越好。若要準確地測定包合率,需要選擇測定包合物總藥量的提取溶劑和未包合藥量的洗脫溶劑。本實驗以川芎油中主要成分藁本內酯和洋川芎內酯A的包合率之和為評價指標,根據藁本內酯和洋川芎內酯A在川芎油中的含量,兩種成分包合率的權重系數分別為0.75、0.25。對包合率測定中幾種常用的溶劑進行考察篩選,確定了準確測定包合率的方法。
藁本內酯包合率Y1=

(1)
洋川芎內酯A包合率Y2=
(2)
包合率=Y1×0.75+Y2×0.25
(3)
2.3.1 包合物提取溶劑的考察
參考文獻方法[13,15],包合物常采用無水乙醇、甲醇、乙酸乙酯、水蒸氣蒸餾等提取其藥物成分。預實驗結果顯示,為確保包合物中藥物成分提取完全,需選用極性大的提取溶劑。按“2.2.3”項下方法操作,保持其他條件不變,分別考察70%乙醇、80%乙醇、95%乙醇、無水乙醇和甲醇對包合物的提取效果,同法回流提取。結果見表1,各溶劑、超聲和回流提取效果均無明顯差異,RSD均小于1%,綜合考慮簡便性和經濟實用性,選擇70%乙醇作為包合物的提取溶劑。
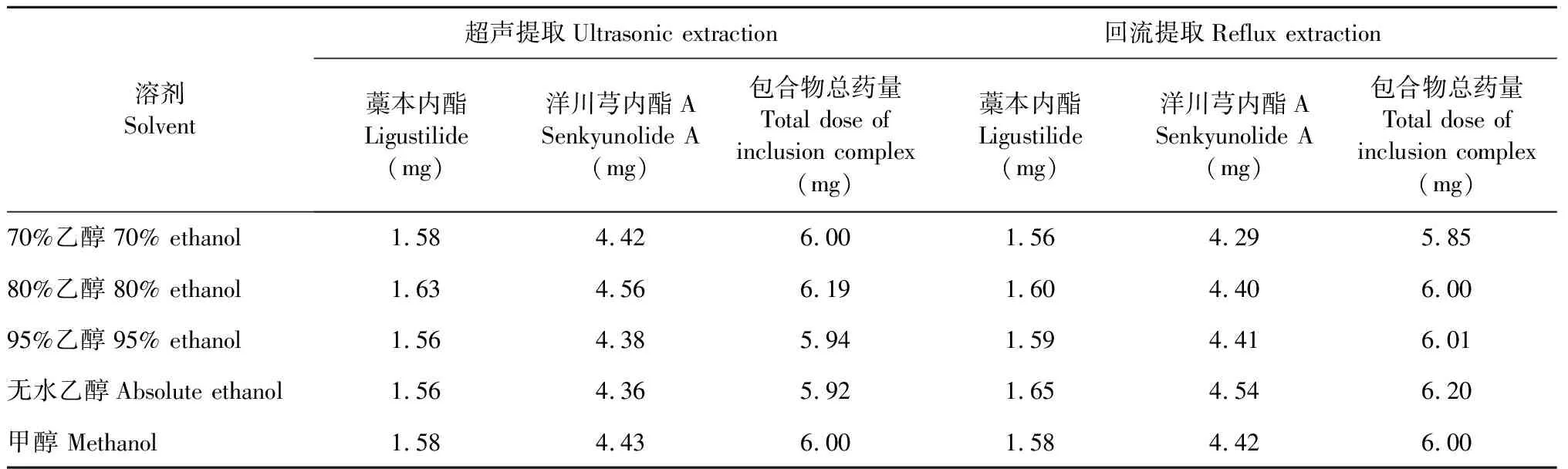
表1 不同溶劑超聲和回流提取結果(n=3)Table 1 Ultrasonic and reflux extraction results by using different solvent (n=3)
2.3.2 包合物洗脫溶劑的考察
參考文獻方法[6,15,16],未包合物的洗滌常使用石油醚、環己烷、乙酸乙酯、乙醇、蒸餾水等。洗脫溶劑需將包合物中未包合的藥物充分洗滌,且不使包合物脫包。預實驗結果顯示,石油醚極性小,能溶解川芎油,且不使包合物脫包,故采用石油醚對未包合物進行洗脫,并對洗脫次數進行考察。
取包合物粉末約0.1 g,精密稱定,置具塞錐形瓶中,加入石油醚25 mL,超聲處理(功率300 W,頻率40 kHz)30 min,放冷,濾過,濾渣用石油醚洗滌3次,每次3 mL,濾液和洗液合并,40 ℃減壓回收石油醚,精密加入95%乙醇10 mL,搖勻,濾過,取續濾液,即得。濾渣再次加入石油醚25 mL,同上操作4次。結果見表2,第3次和第4次提取藥量極低,故采取石油醚超聲提取兩次測定未包合藥量。
表2 石油醚不同洗脫次數提取結果Table 2 Extraction results of petroleum ether with different elution
綜上所述,確定包合率的測定方法:按“2.2.3”項下方法操作,測得包合物總藥量;再按“2.3.2”項下方法操作,石油醚提取兩次,測得未包合藥量。按公式(1)、(2)、(3)計算包合率。
2.4 制備方法篩選
文獻報道HP-β-CD包合揮發油的常見方法主要有水溶液法、研磨法和單相法[10-12],故對該3種方法進行考察,篩選出較為合適的包合物制備方法。預實驗結果顯示,水溶液法在滴加川芎油時,于邊超聲邊攪拌的情況下包合率更高,故將水溶液法改進為水溶液超聲攪拌法。
2.4.1 水溶液超聲攪拌法
按川芎油和HP-β-CD的質量比為1∶10,稱取HP-β-CD溶于2倍蒸餾水中,于邊超聲邊攪拌的條件下,將川芎油(用等量95%乙醇稀釋)緩慢滴加至HP-β-CD水溶液中,混合均勻,磁力攪拌2 h(溫度30 ℃,攪拌速度750 rpm),分別于70 ℃減壓回收、干燥。平行制備3份,平均包合率為84.54%±0.34%。
2.4.2 研磨法[11]
按川芎油和HP-β-CD的質量比為1∶10,稱取HP-β-CD于研缽中,加入少量水研磨至面糊狀,緩慢滴加川芎油(用等量95%乙醇稀釋),繼續研磨45 min,轉移至蒸發皿中,70 ℃減壓干燥。平行制備3份,平均包合率為88.37%±1.58%。
2.4.3 單相法[12]
按川芎油和HP-β-CD的質量比為1∶10,稱取HP-β-CD溶于2倍95%乙醇中,將川芎油滴加至HP-β-CD乙醇溶液中,混合均勻,分別于70 ℃減壓回收、干燥。平行制備3份,平均包合率為91.17%±0.21%。
根據以上結果,單相法制備的包合物的包合率最高,且方法簡便易操作,故選擇單相法制備川芎油HP-β-CD包合物。
2.5 單因素考察
2.5.1 乙醇體積分數
按“2.4.3”項下方法,保持其他條件不變,考察乙醇體積分數80%、85%、90%、95%、100%對包合率的影響。結果見圖2,隨著乙醇體積分數的增加,包合物包合率增加,綜合考慮包合率和成本效益,選擇乙醇體積分數85%、90%、95%進行正交試驗。
2.5.2 投料比
按“2.4.3”項下方法,保持其他條件不變,考察川芎油與HP-β-CD的質量比1∶6、1∶8、1∶10、1∶12、1∶15對包合率的影響。結果見圖2,隨著投料比的增加,包合物包合率增加,綜合考慮包合率和輔料用量,選擇投料比1∶6、1∶8、1∶10進行正交試驗。
2.5.3 HP-β-CD與乙醇的質量體積比
按“2.4.3”項下方法,保持其他條件不變,考察HP-β-CD與乙醇的質量體積比為1∶1、1∶2、1∶3、1∶4、1∶5對包合率的影響。結果見圖2,隨著HP-β-CD與乙醇的質量體積比的增加,包合物包合率增加的幅度較小,綜合考慮包合率和成本效益,選擇HP-β-CD與乙醇的質量體積比1∶1、1∶3、1∶5進行正交試驗。
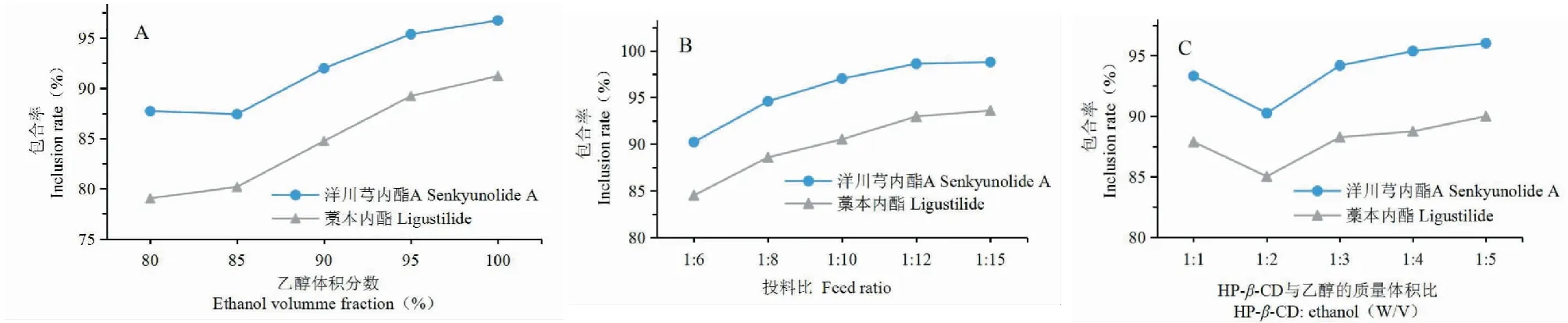
圖2 不同因素對包合率的影響Fig.2 The influence of different factors on the inclusion rate
2.6 正交設計優選包合工藝
考察因素為乙醇體積分數、川芎油與HP-β-CD的質量比、HP-β-CD與乙醇的質量體積比,每個因素選取3個水平,以包合率為評價指標,采用L9(34)正交表安排試驗,試驗設計及結果見表3,方差分析見表4。
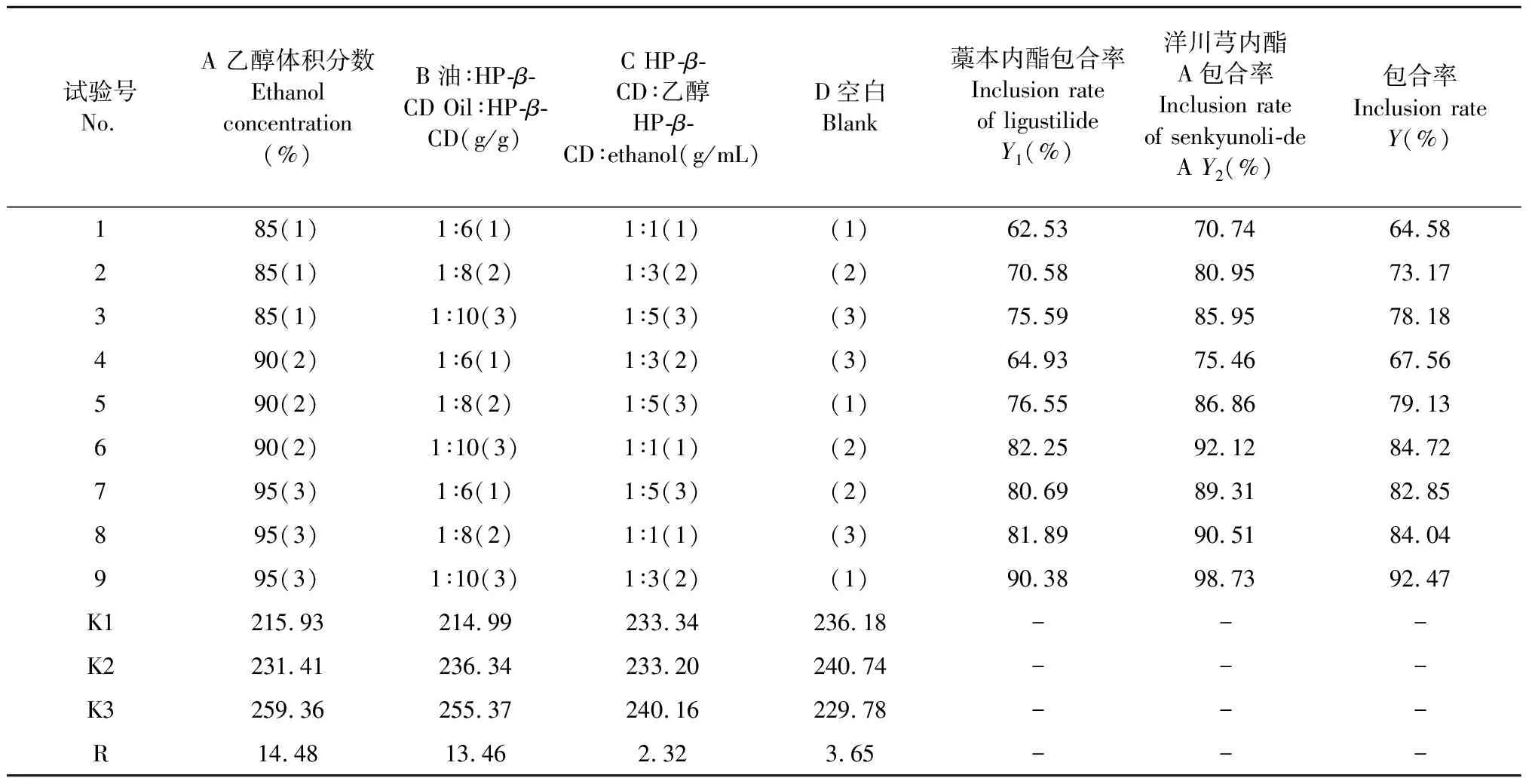
表3 正交實驗設計與結果Table 3 Design and results of orthogonal test

表4 方差分析Table 4 Analysis of variance
由直觀分析可知,各因素對實驗結果的影響大小為A>B>C。方差分析結果表明,A因素存在極顯著性差異(P<0.01),為最重要影響因素;B因素存在顯著性差異(P<0.05),為主要影響因素;C因素無顯著性差異,為次要影響因素。綜合考慮成本與效益,確定最佳制備工藝為A3B3C2,即乙醇體積分數95%,油∶HP-β-CD(g/g)為1∶10,HP-β-CD∶乙醇(g/mL)為1∶3。
按優選的包合工藝制備3批包合物,包合率分別為92.17%、92.62%、92.23%,平均包合率為92.34%,RSD為0.26%,結果重現性好,該工藝穩定可行。
2.7 川芎油包合物的表征
2.7.1 掃描電鏡觀察(SEM)
分別取HP-β-CD、川芎油HP-β-CD物理混合物和川芎油HP-β-CD包合物適量,經導電膠粘樣、噴金鍍膜后,觀察其形貌特征。由圖3可知,HP-β-CD呈中空圓球狀,物理混合物中川芎油呈小顆粒分散在HP-β-CD的表面及四周,而包合物中川芎油成分分子進入到HP-β-CD分子空腔內,使HP-β-CD發生形態學變化,轉呈棱狀,表明包合物形成。

圖3 樣品SEM圖(×500)Fig.3 SEM image of samples (×500) 注:A.HP-β-CD;B.川芎揮發油HP-β-CD物理混合物;C.川芎揮發油HP-β-CD包合物。Note: A.HP-β-CD;B.Chuanxiong Rhizoma volatile oil-HP-β-CD physical mixture;C.Chuanxiong Rhizoma volatile oil-HP-β-CD inclusion complex.
2.7.2 紅外光譜分析(IR)
分別稱取川芎油、HP-β-CD、川芎油HP-β-CD物理混合物和川芎油HP-β-CD包合物適量,其中川芎油采用KBr涂片法,其余采用KBr壓片法,分辨率4 cm-1,光譜范圍4 000 ~400 cm-1全譜掃描,結果見圖4。川芎油的紅外圖譜(A)于1 761 cm-1出現羰基特征峰,為川芎油中內酯類成分;HP-β-CD(B)在3 395 cm-1處出現多締合體羥基的伸縮振動;物理混合物(C)中羰基吸收峰(1 762 cm-1)明顯存在;包合物(D)中羰基吸收峰消失,羥基吸收峰位移至3 412 cm-1,可能與川芎油成分分子進入到HP-β-CD分子空腔中,破壞了其分子內氫鍵有關[17],使得締合羥基向高頻移動。以上波數變化說明包合物已形成。
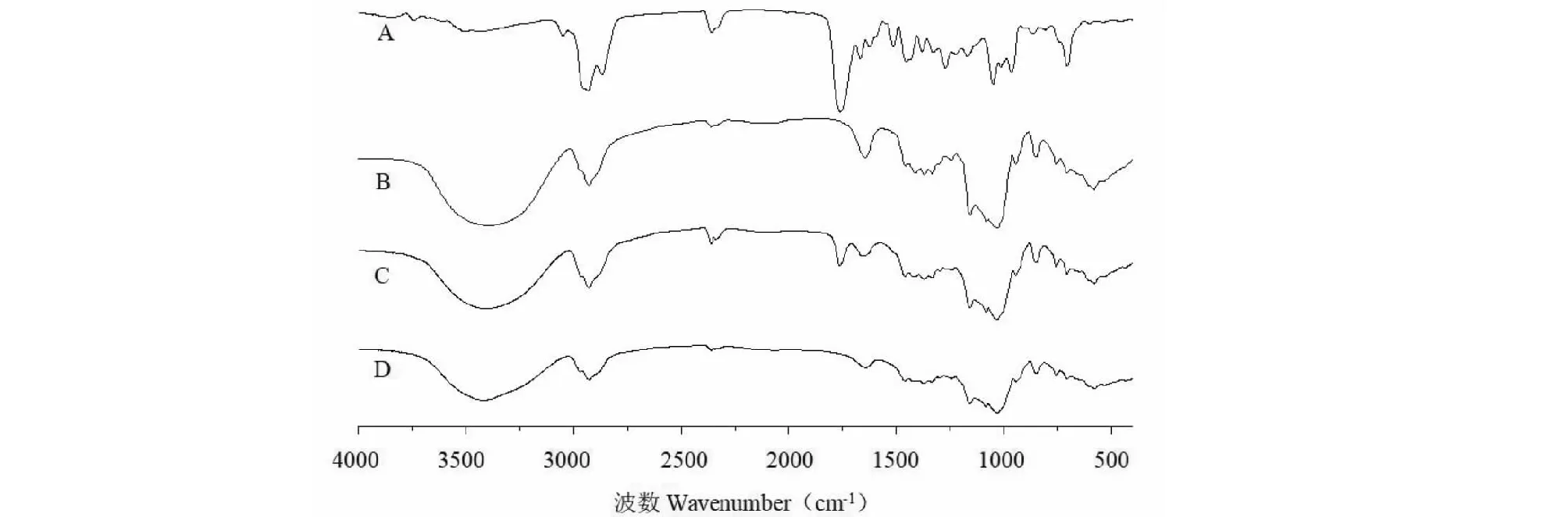
圖4 樣品IR圖Fig.4 IR image of samples 注:A.川芎油;B.HP-β-CD;C.川芎揮發油HP-β-CD物理混合物;D.川芎揮發油HP-β-CD包合物。Note: A.Chuanxiong Rhizoma volatile oil;B.HP-β-CD;C.Chuanxiong Rhizoma volatile oil-HP-β-CD physical mixture;D.Chuanxiong Rhizoma volatile oil-HP-β-CD inclusion complex.
2.8 川芎油HP-β-CD包合物的性能研究
2.8.1 相溶解度法
稱取適量HP-β-CD,加蒸餾水分別配制為0、10、20、30、40、50、60 mmol/L的溶液各10 mL,分別加入過量川芎油,于35 ℃,120 rpm氣浴恒溫振蕩器振搖48 h。待溶液平衡,常溫條件下12 000 rpm離心10 min,取上清液,濾過,取續濾液,按“2.2.1”項下色譜條件測定。以HP-β-CD濃度X(mmol/L)為橫坐標,藁本內酯和洋川芎內酯A的濃度Y(mmol/L)為縱坐標作圖,繪制相溶解度曲線,見圖5。對曲線進行線性回歸,得回歸方程。根據Higuchi和Connors提出的理論[18],按公式(4)、(5)計算增溶倍數和平衡常數K,增溶倍數結果見表5。
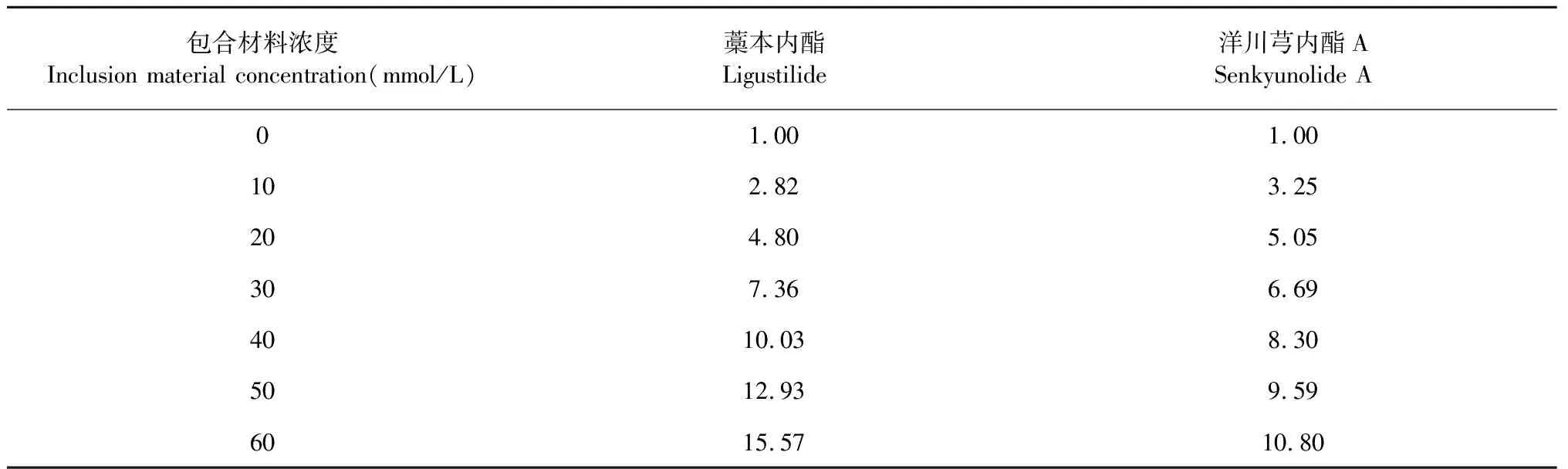
表5 不同濃度HP-β-CD對川芎油中藁本內酯和洋川芎內酯A的增溶倍數(T=35 ℃)Table 5 The solubilization multiples of different concentrations of HP-β-CD onLigustilide and Senkyunolide A in Chuanxiong volatile oil (T=35 ℃)
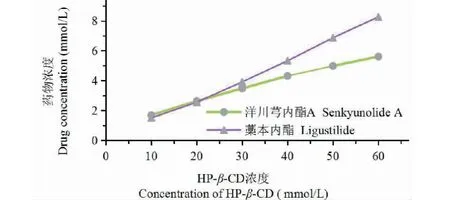
圖5 川芎油HP-β-CD相溶解度圖Fig.5 Chuanxiong Rhizoma oil HP-β-CD phase solubility diagram
由相溶解度曲線判斷,在試驗選取的濃度范圍內,藁本內酯、洋川芎內酯A濃度隨著HP-β-CD濃度呈線性增長,包合類型為AL型,可形成摩爾比為1∶1的可溶性包合物,得到的回歸方程分別為Y= 0.137 8X+ 0.531 1(R2= 0.997 1),Y= 0.078 7X+ 0.519 5(R2= 0.994 3)。包合物中藁本內酯和洋川芎內酯A的平衡常數K分別為300.93、164.42,說明形成的包合物較穩定,HP-β-CD對川芎油有較好的包合效果。在試驗選取的濃度范圍內,藁本內酯和洋川芎內酯A的最大增溶倍數可達15.57、10.80。
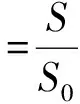
(4)
(5)
式中S為川芎油在HP-β-CD溶液中的溶解度,S0為川芎油在水中的飽和溶解度。
2.8.2 加速熱穩定性試驗
川芎油在儲存過程中不穩定,溫度、氧氣、pH和光照均對其穩定性有影響,溫度為主要影響因素[19,20]。故對包合物的熱穩定性進行考察。
分別稱取適量川芎油、HP-β-CD物理混合物和HP-β-CD包合物于密閉潔凈容器中,攤成≤5 mm厚的薄層。置于60 ℃恒溫培養箱中,考察10天,于第0、5、10天取樣,按“2.2.3”項下方法制備供試品溶液,計算樣品與第0天的相對保留率。結果見表6,HP-β-CD包合物相比物理混合物和川芎油的穩定性有顯著性改善。
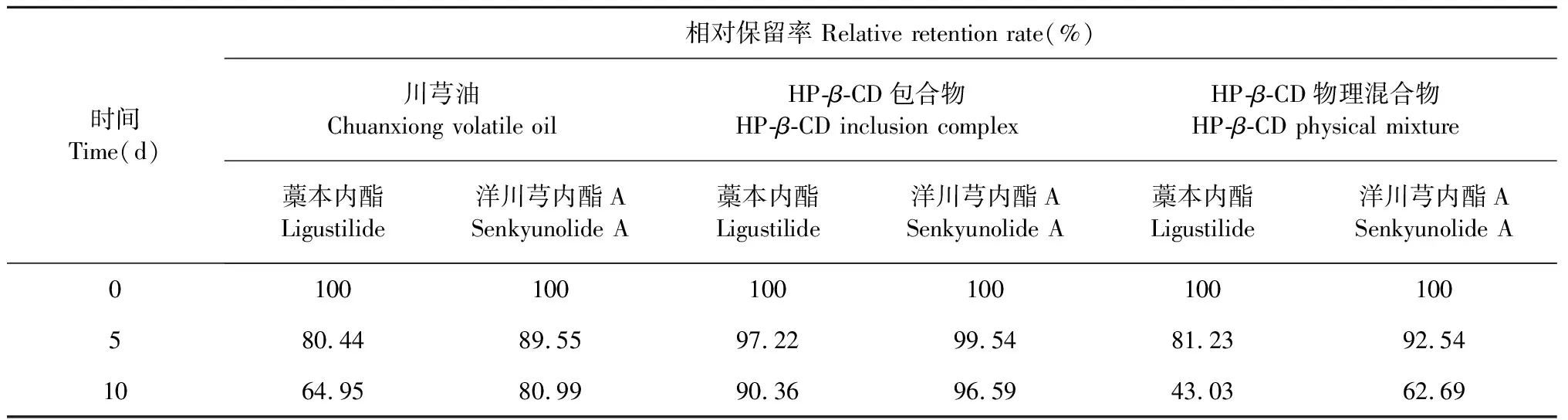
表6 川芎油包合物熱穩定性試驗結果Table 6 Thermal stability test results of Chuanxiong Rhizoma oil inclusion complex
3 討論
本實驗采用單相法進行包合,川芎油一次性加入HP-β-CD的乙醇溶液中,且無需研磨、攪拌或超聲處理,直接減壓回收、干燥即得包合物。該方法簡便、實用、收率高,適合于工業化大生產。
藁本內酯和洋川芎內酯A尚無法定的含量測定用對照品,且不穩定,對照品純度標定和保存困難,故本課題組前期建立了以丁苯酞為對照品,一測多評法測定川芎油中藁本內酯和洋川芎內酯A的含量,該方法簡便可行[14]。本文對含量測定方法學進行了考察,專屬性、線性、精密度、穩定性和重復性等符合定量分析的要求。
關于包合率的測定,文獻報道一般以洗滌后的包合物測得的藥物量除以實際投入量為包合率。洗滌操作常在制備包合物過程中用水或有機溶劑淋洗抽濾后的濾餅,或采用有機溶劑萃取包合物溶液[6,9,10,16],二者均會使包合物脫包,測得包合率偏低。此外,另有文獻報道采用水蒸氣蒸餾法[9,10,12,16]提取包合物中的揮發油,該方法揮發油定量提取不完全,導致測得包合率偏低,且重現性差。本實驗經比較研究,采用極性大的70%乙醇提取測定包合物總藥量,再以極性小的石油醚提取測定未包合藥量,包合物總藥量減去未包合藥量即為真實的包合物藥量,該方法能更準確地測定包合率。
HP-β-CD相溶解度曲線在一定濃度范圍內呈線性增長,藁本內酯斜率大于洋川芎內酯A,表明HP-β-CD對藁本內酯的增溶效果更好。此外,本實驗對川芎油β-CD包合物也進行了相溶解度的考察,在相同的試驗濃度范圍內,其相溶解度曲線呈非線性增長,藁本內酯和洋川芎內酯A的增溶倍數分別為2.36、2.95,遠低于HP-β-CD,與文獻報道一致[21],表明HP-β-CD包合物具有明顯的優勢。川芎油HP-β-CD包合物的生物利用度及藥理學活性等有待進一步實驗研究。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
