基于絲束電極的環氧涂層失效演變行為研究
2021-05-17 01:57:18孫祥太
電鍍與精飾 2021年4期
孫祥太
(中石化中原油田分公司,河南濮陽457001)
金屬腐蝕問題不僅會帶來大量金屬損失,也會引起諸多的安全隱患[1‐3],金屬防護一直是各國學者爭相研究的熱點。有機涂層防護技術廣泛應用于金屬防護領域,從最初的石油瀝青防腐層到如今的氟碳漆等多種涂層,而有機環氧涂層以其較高的交聯密度、高強度等優點廣泛應用于金屬防護領域[4‐7]。目前,各國學者研究的熱點集中在以有機環氧涂層為基礎,添加多種物質,形成諸如富鋅環氧涂層、石墨烯涂層、碳納米管涂層等多種高性能的有機涂層[8‐10]。但對于不同涂層來說,腐蝕介質擴散引起的涂層失效過程到目前為止仍然沒有統一定論,尤其是在涂層失效過程中的失效位點確定研究較少[11‐12]。絲束電極技術在局部失效位點的確定、表面動力學參數變化趨勢的研究等方面具有巨大的優勢[13],因此本文以DGEBA 環氧涂層為研究對象,通過絲束電極技術、電化學測試技術和圖像表征技術研究了涂層失效的全過程,以期為環氧涂層改性等研究提供借鑒。
1 實驗部分
1.1 絲束電極制備與表面參數測試
絲束電極制備:以透明有機玻璃板為基礎,采用精雕機加工10×10 的陣列圓孔,孔徑為2 mm,小孔之間的間距為0.5 mm。打磨至光亮的直徑為2 mm的X80 鋼絲插入到小孔內并與有機玻璃板平行,然后采用環氧樹脂對陣列電極進行封裝,保證鋼絲之間完全絕緣。環氧樹脂室溫固化72 h后打磨陣列電極,使之完全漏出X80鋼金屬基體,最后通過金相砂紙打磨電極至平整。將打磨后的絲束電極用水清洗,然后放入真空干燥箱中備用。
涂層制備:實驗涂層采用高分子DGEBA 環氧涂層(雙酚A 縮水甘油醚),手工噴涂至絲束電極表面,室溫固化168 h 后,采用五點測試法確定其涂層厚度為10±2 μm。
電位和電流密度數據采集:編寫Labview 程序,通過數據采集卡實現不同實驗時間條件下的絲束電極電位和電流密度值的自動采集及保存,實驗周期為72 h。
1.2 涂層表面圖像分析
涂層體系制備:以尺寸為 25 mm × 25 mm × 2 mm 的X80鋼試片為基體,依次采用丙酮除油、去離子水清洗和酒精除水后,干燥放入真空干燥箱中備用。使用前,通過環氧樹脂封裝,只留下一個面積為25 mm × 25 mm 的工作面。實驗前,采用金相砂紙打磨至鏡面,手工涂覆環氧涂層,室溫固化168 h 后用于浸泡實驗。
圖像分析:通過掃描電鏡觀察涂層的表面形態,同時除掉涂層后觀察金屬表面的腐蝕形態。
1.3 實驗條件
采用分析純NaCl 和去離子水配制質量分數為3%的NaCl 實驗溶液,整個實驗裝置放置于恒溫恒濕箱中,實驗溫度設置為25 ℃。
2 結果與討論
2.1 環氧涂層表面電位和電流密度分布
水分子在涂層中的擴散過程取決于兩個方面:(1)由于涂層本身固化劑的揮發,在涂層內部存在大量的自由體積,這就是在初始時刻腐蝕性介質(水)發生擴散的重要途徑[14];(2)由于涂層本身交聯結構的不均勻性,涂層本身高分子鏈對水分子擴散過程的阻擋作用是不一致的[15‐16]。因此,水分子在涂層中擴散必然會存在一個失效位點,該點可以認為是水分子第一個到達涂層/金屬界面的位置。
圖1 為實驗3 h 時環氧涂層表面電位分布,圖2為環氧涂層表面電位和電流密度分布隨實驗時間的變化情況。可以看出,實驗進行到第3 h 時,環氧涂層表面電位最正值出現在(2,8)位置,為-0.691 V,最負位置出現在(9,4)、(9,5)和(9,9)位置,為-0.708 V。因此可以判斷出此時坐標點(2,8)位置水分子已經到達涂層/金屬界面,三個電位最負位置涂層仍然表現出完好的防護特征。其他絲束電極位置的相對于初始時刻的涂層體系(腐蝕電位為-0.708 V),均發生不同程度的正向偏移,這說明水分子在涂層中的擴散過程已經展開,并且電位正向偏移程度越大,水分子擴散程度越高。隨著實驗的進一步進行,當時間到達6 h 時,涂層表面的電位分布更加不均勻,最大電位差達到34 mV,與3 h 的電位分布圖像相比,陽極區數量基本保持不變,而陰極區的電位值大部分發生正向偏移,局部位置發生負向偏移,大陰極、小陽極的分布逐漸趨向于均衡。當實驗時間到達12 h 時,多處絲束電極位置水分子已經到達涂層/金屬界面,同時陰極區電位發生大幅度正向偏移。
相關研究表明,在涂層失效初期,即局部位置水分子到達涂層金屬界面上時,形成的“大陰極、小陽極”能夠極大地促進局部腐蝕,此時在涂層界面上,首先以金屬的活化腐蝕過程為主,當界面上的氧氣消耗殆盡,此時由于界面的封閉性,形成的氧濃差電極繼續促進局部的點蝕過程。因此,當浸泡時間由3 h增大到6 h時,坐標(2,8)位置電位值持續向正向偏移,同時陰極/陽極的最大電位差最大。當時間達到24 h 時,整個涂層體系的電位發生大幅度的負向偏移,表面電位分布范圍為-0.722~-0.738 V,同時在后續的48 h里,涂層體系表面電位基本保持不變。
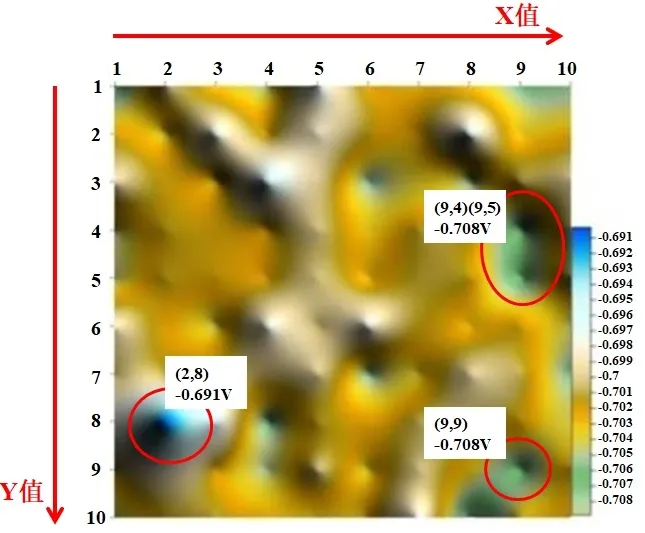
圖1 實驗3 h時帶涂層絲束電極表面電位分布Fig.1 The potential distribution on the surface of the coated wire bundle electrode at experiment time of 3 h

圖2 環氧涂層表面電位和電流密度分布隨實驗時間的變化規律Fig.2 The variation of surface potential and current density distribution of epoxy coating with experiment time
結合6 h 和12 h 圖像分析可得,當實驗時間達12 h 時,水分子已經大量達到涂層/金屬界面上,界面的電化學腐蝕過程全面展開,此時界面上的大量腐蝕產物向涂層內部擴散,堵塞水分子的擴散通道,腐蝕性介質難以繼續到達涂層/金屬界面上,因此整個涂層體系在隨后的時間里電位發生大幅度負向偏移,并且在長時間里保持不變。但是事實上,根據現場實驗結果發現,雖然此時涂層體系處于穩定狀態,并且表現為較好的防護特征,但是由于界面上電化學反應導致腐蝕產物積聚導致涂層與金屬之間的結合力降低,促進了環氧涂層的剝離。
涂層體系表面電流密度分布表現為相同的變化規律。隨著實驗的進行,涂層體系出現明顯的陰極和陽極,其中陽極區域即為水分子到達涂層/金屬界面的區域。當實驗進行到6 h 時,與3 h 的表面電流密度分布相比較,此時陰/陽極最大電位差顯著增大,并且陽極區和陰極區的電流密度也顯著增大,這是由于整體陰極區和陽極區整體趨于均衡造成的。當實驗進行到24 h 時,此時表面電流密度分布相對更加均勻,并且在后續的48 h里基本保持不變,這是由于涂層/金屬界面上腐蝕產物堵塞擴散通道導致的。
圖3 為涂層體系表面陰/陽極電位差和平均電流密度值隨時間的變化趨勢。從涂層表面電位和電流密度分布隨時間的變化規律上可以看出,涂層的失效分為兩個階段:水分子到達涂層金屬界面形成擴散通道,界面上的腐蝕產物向涂層中擴散堵塞擴散通道。在整個過程中,涂層體系陰/陽極電位差和平均電流密度均隨著實驗時間延長而減小,這說明涂層的失效首先發生在水分子到達涂層/金屬界面的位置,形成了大陰極、小陽極的局部腐蝕特征。隨后當大量水分子到達涂層/金屬界面,涂層表面電位和電流密度分布均趨于均勻,表示涂層的全面失效。
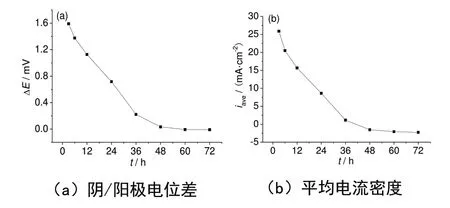
圖3 陰/陽極電位差和平均電流密度值隨時間的變化趨勢Fig.3 The variation of the potential difference of anode and cathode and the average current density with experiment time
2.2 涂層表面特征分析
開展涂層樣品的浸泡實驗,其中浸泡時間為3 h的涂層表面形貌測試結果如圖4 所示。浸泡時間為12 h的涂層表面形貌及去除掉防腐層后金屬表面的腐蝕形貌測試結果也列于如圖4。由圖4可以看出,當浸泡時間為3 h時,涂層表面形貌相對平整、均勻,局部位置凸起不明顯。從表面形貌的局部放大圖像可以看出,部分位置顏色由黑色轉變為白色。該位置處與周圍形貌有明顯不同,發生了局部厚度增大現象導致該處呈現為亮白色,表明此時水分子發生明顯的擴散,可能已經到達涂層/金屬界面,電化學反應的發生導致了涂層厚度的變化。
當浸泡時間為12 h時,涂層表面狀態與3 h樣品測試結果相比無明顯變化。從前文分析可知,當浸泡時間到達12 h時,水分子已經大量到達涂層/金屬界面,界面的電化學腐蝕過程全面展開。除掉表面涂層后觀察金屬表面腐蝕特征,表現為明顯的點蝕特征。因此,在涂層失效前期,水分子到達涂層/金屬界面導致的基體腐蝕以點蝕為主,與前文分析結果相吻合。

圖4 不同浸泡實驗時間的涂層表面形態和金屬表面腐蝕形貌Fig.4 Coating surface morphology and metal surface corrosion morphology of different immersion ex‐periment time
3 結論
(1)涂層失效起始于失效位點,即水分子優先達到涂層/金屬界面的位置,此時整個涂層體系形成明顯的陰極和陽極。隨著實驗時間的推移,大量水分子進入導致陰極區和陽極區分化明顯。當涂層/金屬界面上電化學腐蝕過程全面展開,界面腐蝕性介質向涂層中擴散,阻塞擴散通道,整個涂層體系電位大幅度負向偏移,涂層發生剝離。
(2)在涂層失效前期,在失效位點處形成了大陰極、小陽極的腐蝕原電池,同時氧濃差電池的形成也促進了局部點蝕特征。
猜你喜歡
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
當代陜西(2020年13期)2020-08-24 08:22:02
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
制造技術與機床(2017年5期)2018-01-19 02:49:17
金秋(2017年4期)2017-06-07 08:22:16
中國材料進展(2016年10期)2016-12-26 06:50:20
濰坊學院學報(2016年2期)2016-12-01 13:00:11
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
