慢性阻塞性肺病藥物治療靶點及其藥物研發進展
2021-05-08 08:31:42葉連寶陳偉強
中國藥科大學學報 2021年2期
關鍵詞:研究
顏 佩,葉連寶,陳偉強
(1廣東藥科大學藥學院,廣州510006;2廣東藥科大學護理學院,廣州510006)
慢性阻塞性肺病(chronic obstructive pulmo?nary disease,COPD)是全球高發病率和高病死率的疾病之一,與呼吸道的慢性炎癥增強有關[1]。COPD患者因肺部病變或肺內氣道狹窄導致呼氣和吸氣時氣流受限,易出現呼吸困難的癥狀,一般用氣流受限(airflow limitation,AL)即伴有小氣道阻塞和用力呼氣容積(forced expiratory volume,FEV)較低來定義,1 s用力呼氣容積占用力肺活量百分比(FEV1/FVC)是評價AL的一項敏感指標,當FEV1/FVC<0.7時被定義為AL。慢性支氣管炎和肺氣腫患者出現氣流受限且不能完全可逆時即可診斷為COPD[2]。此外,COPD通常伴隨肺功能的異常下降,由此而導致的慢性呼吸衰竭是該病的主要死亡原因。本文重點綜述了COPD的發病機制、治療靶點以及相關藥物的最新臨床研究進展,以期為COPD靶向藥物的開發提供參考。
1 COPD的致病因素
吸煙通常被認為是COPD最主要的致病因素[3],然而多項研究表明部分患者沒有吸煙史[4]。此外環境誘因如煙草煙霧、空氣污染、汽車尾氣以及有毒氣體等在COPD的發展中也發揮著十分重要的作用[5]。其他因素還包括性別和年齡:一般來說,由于男性吸煙人數多于女性,男性COPD的患病率通常高于女性,但實際上女性患COPD的風險更高[6]。COPD的病死率與年齡呈正相關,老年患者更容易并發呼吸衰竭等而導致死亡[7]。此外,支氣管高反應性和哮喘導致的肺功能下降,遺傳、社會經濟地位、貧困、體重指數和呼吸系統感染等多種因素都與COPD密切相關[8]。弄清楚COPD患病的危險因素有助于COPD的預防、篩查和治療,同時也為靶向COPD治療藥物的發現奠定基礎。
2 COPD的發病機制
2.1 氧化應激
氧化物和抗氧化物失衡會促進炎癥基因的表達、氣道黏液分泌以及抗蛋白酶失活[9]。核因子E2相 關 因 子2(nuclear factor E2-related factor,Nrf2)通過介導Keap1-Nrf2-ARE等信號通路能調節抗氧化基因的表達,通常情況下處于未激活狀態的Nrf2與Keap1在細胞質中結合,長時間未激活的Nrf2會被泛素化,進而被降解,阻止其在細胞質中累積。當被活性氧(reactive oxygen species,ROS)刺激時,Nrf2從Keap1-Nrf2的結合態中游離而被釋放出來,進而轉移到細胞核中與ARE結合,繼而激活下游目標基因促使其轉錄。因此,靶向該信號通路的作用環節可能成為治療COPD的潛在靶點。
2.2 蛋白酶/抗蛋白酶失衡
肺中性粒細胞浸潤通過釋放蛋白酶如中性粒細胞彈性蛋白酶(neutrophil elastase,NE),組織蛋白酶G(cathepsin-G,CG),蛋白酶3(proteinase 3,PR3)而導致蛋白酶和抗蛋白酶失衡[10],這一過程在COPD的發生中發揮著關鍵作用。抗蛋白酶在細菌包裹、病原菌吞噬、黏液素分泌亢進和黏液纖毛清除中起重要作用。同時NE通過降解肺實質結締組織的主要成分彈性蛋白而加劇COPD。若彈性蛋白酶活性增加或抗蛋白酶活性降低和功能缺乏時會導致肺氣腫和中性粒細胞炎癥及黏液分泌增多,進而可能發展成不可逆的COPD。
2.3 免疫機制
與COPD相關的免疫機制主要包括先天免疫激活、適應性免疫的轉變和氣道上皮的形態學改變。先天免疫激活是指吸入的顆粒和病原體被肺泡上皮細胞和巨噬細胞質膜上的受體(pattern recognition receptors,PRRs)識別,通過caspase-1依賴機制產生炎性細胞因子,形成與核苷酸結合的寡聚結構域樣(NLR)P3炎性小體。Pauwels等[11]研究發現,吸煙誘導的肺部炎癥與NLRP3炎癥小體或caspase-1的缺陷無關,但目前多項研究支持NLRP3炎性小體參與COPD的發病機制,這一領域仍存在爭議,需要進一步研究。適應性免疫的轉變是細胞和組織損傷釋放的抗原被樹突狀細胞識別,并呈遞給T淋巴細胞,從而激活適應性免疫。煙霧顆粒誘導的NETs可增強漿細胞樣樹突狀細胞,產生CD4陽性T細胞Th1和Th17[12],進而激發適應性免疫的轉變。氣道上皮的形態學改變是指當慢性接觸煙霧顆粒時會直接影響氣道上皮的形態和功能。有害顆粒介導的ROS通過表皮生長因子受體依賴機制破壞上皮屏障,同時也激活RHO1相關的蛋白激酶,最終導致E-cadherin基因表達的減少、黏附連接的破壞、上皮屏障的破壞和E-cad?herin表達的減少進而誘導上皮-間質轉化,導致MMPs和生長因子的產生異常、氣道破壞和重塑[13-14]。COPD的免疫機制十分復雜,很多研究處于初步階段,還需繼續深入探究。
2.4 細胞衰老和細胞修復機制
COPD的發病率與年齡增長呈正相關主要有以下幾個原因,如端粒縮短、細胞衰老、激活磷脂酰肌醇-3激酶(phosphatidylinositol-3-kinase,PI3K)/哺乳動物雷帕霉素靶蛋白(mammalian target of rapa?mycin,MTOR)信號通路、DNA修復缺陷、microRNA模式異常和表觀遺傳改變等[15]。其潛在的作用機制可能是細胞內外ROS的產生,ROS水平過高時,p53腫瘤抑制蛋白被激活導致細胞周期阻滯和衰老[16]。PI3K/AKT/mTOR信號通路是加速細胞衰老導致COPD的主要原因。正常情況下肺具有再生能力,由氣道基底祖細胞分化而來的新上皮細胞替代損傷的氣道上皮細胞,當肺基底祖細胞數量減少,喪失了自噬能力和多潛能,影響了細胞自我修復能力,從而可能引發COPD,基底祖細胞的功能障礙與COPD嚴重程度相關[17]。
2.5 細胞壞死和細胞自噬
細胞壞死是一種精心設計的細胞死亡形式,受體相互作用于依賴性蛋白激酶(RIPK)3和非依賴性caspase的程序性壞死被定義為壞死性凋亡[18]。Pouwels等[19]研究發現煙霧誘導的壞死性凋亡和DAMPs的釋放在模型小鼠中引發嗜中性粒細胞炎癥,細胞壞死抑制劑necrostatin-1可以使炎癥減輕。Wang等[20]研究報道內質網伴侶蛋白GRP78能促進煙霧誘導的炎癥反應和氣道上皮細胞黏液增生,其作用機制可能是上調壞死性凋亡以及激活NF-κB和激活蛋白1(AP-1)。自噬被認為是一種非選擇性的自我降解系統,且具有選擇性,包括對特定細胞器和病原體的選擇性降解。有報道稱,自噬可選擇性地靶向線粒體(線粒體吞噬),與正常人相比COPD患者肺中線粒體自噬標志物蛋白水平降低,即通過抑制線粒體吞噬作用,增強氣道上皮細胞線粒體ROS的產生并促進細胞衰老[21]。這些研究表明細胞壞死和細胞自噬在COPD的發生發展中起著至關重要的作用。
3 COPD治療藥物的臨床研究進展
3.1 支氣管擴張藥物
3.1.1 β2受體激動劑 經吸入給藥的支氣管擴張劑是治療COPD的一線藥物,直接作用于氣道以減少全身副作用。β2受體激動劑通過激活氣道平滑肌細胞上的β2-受體進而松弛氣道平滑肌、擴張支氣管[22]。部分藥物已經批準用于COPD的臨床治療或正進行臨床試驗,包括短效β2受體激動劑(shortactingβ2-adrenoreceptor agonists,SABAs),長效β2受體激動劑(long-actingβ2-adrenoreceptor agonists,LABAs)。目前FDA批準用于治療COPD的SABA及LABA藥物具體情況如表1所示。這里主要討論β2受體激動劑的最新研究進展,及部分代表藥物的臨床研究情況。其中沙美特羅(salme?terol)、維蘭特羅(vilanterol)、阿貝替羅(abediterol)屬于水楊醇類LABA藥物,是目前被研究的最為深入的一類β2受體激動劑。川丁特羅(trantinterol)是一種新型的LABA藥物,其特有的親脂性結構使其作用時間長于其他藥物,目前該藥正在進行哮喘治療的Ⅳ期臨床實驗[23]。
3.1.2 毒蕈堿拮抗劑 毒蕈堿拮抗劑作用于迷走神經膽堿能分支,通過降低膽堿能張力放松氣道平滑肌,減少空氣滯留和勞力性呼吸困難,改善氣道狹窄,膽堿能M3受體在介導平滑肌收縮方面具有重要的臨床意義[24]。部分作用于該受體藥物已批準用于臨床治療或正進行臨床試驗,包括短效毒蕈堿拮抗劑(short-acting muscarinic antago?nists,SAMAs)以及長效毒蕈堿拮抗劑(long-acting muscarinic antagonists,LAMAs)。目前FDA批準用于治療COPD的SAMA,LAMA類藥物具體情況如表1所示。這里主要討論毒蕈堿拮抗劑的最新研究進展,及部分代表藥物的臨床研究情況。由Theravance Biopharma公司研發的一種新型LAMA藥物瑞維那新(revefenacin)對M3受體具有較高親和力(Ki=0.18 nmol/L),其平行組高中低3個劑量(88,175,350μg)進行為期28 d的Ⅱ期臨床研究,結果顯示超過80%的患者FEV1較基線升高至少100 mL,支氣管持續擴張24 h,這一結果得到了為期12個月的Ⅲ期臨床安全性研究的支持[25],于2018年11月被FDA批準用于COPD的治療。BCQB是一種新型的SAMA,其化學結構與LAMA藥物相似,BCQB對M3受體有高親和力(p Ki=8.21)且對M2受體有選擇性,有望成為一種新型毒蕈堿拮抗劑治療COPD。

表1 FDA批準用于治療慢性阻塞性肺病(COPD)的支氣管擴張藥
3.1.3 作用于β2受體和毒蕈堿受體的雙靶點藥物 MABA是一類新型具有毒蕈堿拮抗劑和β2激動劑雙重作用的雙靶點藥物。由GlaxoSmithKline公司研發的巴芬特羅(batefenterol)是最新的MABA類藥物,Ⅱ期臨床研究結果顯示該藥的所有測試劑量的耐受性優于比安慰劑和陽性對照沙美特羅(50μg,每天2次),最佳劑量是400μg每天1次或200μg每天2次,能顯著改善中度至重度COPD患者的FEV1。AstraZeneca公司研發的AZD8871在Ⅱ期臨床研究結果顯示,當給予劑量為400或1 800μg時,可以使COPD患者支氣管持續擴張36 h,給藥兩周后FEV1顯著增加,且在100/600μg劑量下無嚴重不良事件發生。LAS190792是一種新型長效MABA,吸入該藥可以誘導犬支氣管持續擴張13.3 h,且心臟效應較小,在Ⅰ期臨床研究階段中幾乎沒有副作用。目前處于臨床研究階段的MABAs藥物見表2。
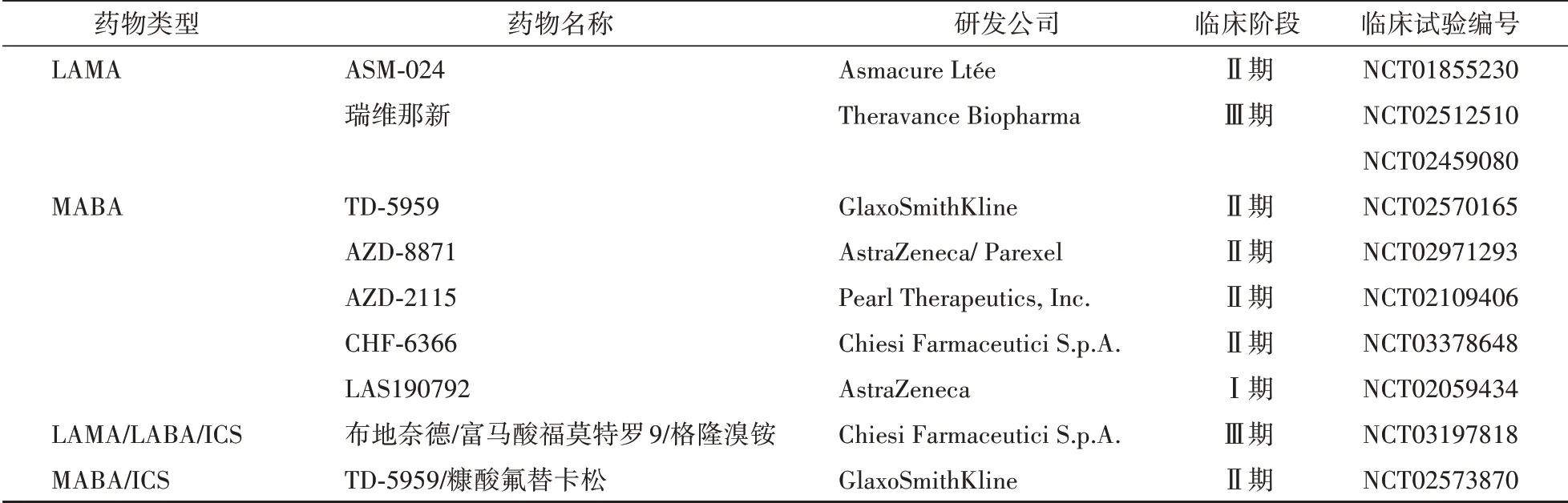
表2 用于COPD的支氣管擴張藥和支氣管擴張藥/ICSFDCs的臨床研究
3.1.4 LAMAs和LABAs藥物的聯合應用 LAMA/LABA復方制劑(fixed-dose combinations,FDCs)具有擴張支氣管和抗炎的雙重作用,藥物的聯合使用有助于減少副作用并增加療效。近年來,新型支氣管擴張劑的發現主要集中在LAMA/LABA FDCs或支氣管擴張劑與吸入皮質類固醇(inhaled cortico?steroid,ICS)的聯合使用。聯合用藥的Ⅲ期臨床實驗結果顯示QVA149(茚達特羅(indacaterol)/格隆溴銨(glycopyrroniumbromide),LABA/LAMA FDCs)比LABA/ICSFDCs(沙美特羅(salmeterol)/氟替卡松(fluticasone))對患者FEV1改善更明顯。因此,LABA/LAMA/ICS三聯吸入治療可能更利于改善肺功能,防止病情加重。新近聯合使用藥物臨床研究情況如表2,上市批準情況如表3。

表3 最新批準用于COPD的LAMA/LABA FDCs和支氣管擴張劑/ICSFDCs
3.1.5 茶堿及其衍生物 茶堿具有支氣管擴張的作用,因此被廣泛用于治療各種呼吸道疾病。低劑量茶堿可增加COPD患者肺泡巨噬細胞HDAC2的表達和活性,從而逆轉皮質類固醇的抵抗作用[26]。多索茶堿的抗炎、擴張支氣管作用與茶堿相當,但與茶堿相比對COPD患者肺功能的改善更有效,副作用更少。除了茶堿和多索茶堿外,還有300多種茶堿衍生物(例如氨茶堿和葉黃素),其中許多藥物對ARs和PDEs沒有選擇性,可能會引起副作用。因此,設計和合成無PDE抑制作用的新型茶堿衍生物可能為COPD治療提供新的機會。

3.2 具有抗炎作用的靶點藥物
3.2.1 靶向炎癥介質的藥物 靶向炎癥介質的藥物主要有以下幾類:細胞因子抑制劑和趨化因子受體拮抗劑,含有NOD-、LRR-和pyrin結構域蛋白3(NOD-,LRR-and pyrin domain-containing 3,NLRP3)炎性體抑制劑,白三烯B4(leukotriene B4,LTB4)受體拮抗劑和LTB4合成調節劑。細胞因子和趨化因子是治療炎性氣道疾病的重要靶標,目前研究較多的是CXCR2拮抗劑,根據結構特點可以分為甲酰胺類(SCH527123)、嘧啶類(AZD-5122,AZD-5069)、二芳基脲類(GSK1325756)、噻唑嘧啶類(AZD-8309),由默沙東研發的那伐立辛(navarixin)是一種強效的CXCR2拮抗劑,在為期12周口服治療的臨床Ⅱ期研究中發現,該藥對COPD患者FEV1有改善作用,但該藥于2018年12月終止了臨床研究。一種二芳基脲類CXCR2拮抗劑SB-656933可影響臭氧誘導的氣道炎癥[27],目前正處于Ⅰ期臨床研究。另一種二芳基脲類CXCR2拮抗劑GSK1325756在Ⅱ期臨床研究也有一定進展。阿斯利康研發的一種嘧啶類CXCR2拮抗劑AZD-5122正處于Ⅰ期臨床研究,另一種嘧啶類CXCR2拮抗劑AZD-5069針對COPD患者開展的Ⅱ期臨床研究已于2011年完成,可后未見更多進展。CCR1拮抗劑AZD-4818Ⅱ期臨床研究發現其對中度至重度COPD患者無效。NLRP3炎性體抑制劑通過間接阻斷細胞因子IL-1β和IL-18而起作用,目前這一靶點藥物對COPD開展的臨床研究非常有限,僅幾個報道與COPD相關,如MCC950、SKF-39162、TAK-242、Bromoxone。這些抑制劑在COPD的治療中要么缺乏臨床研究,要么臨床研究結果不容樂觀,目前大都處于終止狀態。值得慶幸的是還有5-LO抑制劑PEP03正處于Ⅱ期臨床研究中,但具體研究情況并不明確。靶向炎癥介質藥物的臨床研究情況見表4。

表4 用于COPD的趨化因子受體拮抗劑臨床研究

3.2.2 蛋白酶抑制劑 靶向蛋白酶藥物有以下幾類:中性粒細胞彈性蛋白酶抑制劑(neutrophil elastase inhibitors,NEI)、基質金屬蛋白酶(matrix metalloproteinase,MMP)抑制劑、蛋白酶3(protein?ase 3,PR3)抑制劑。人中性粒細胞彈性蛋白酶(human neutrophil elastase,HNE)在炎癥疾病的治療領域是一個十分有希望的靶點,近幾年HNEI在COPD的治療研究較多。西維來司他(sivelestat)是唯一批準上市的HNEI,用于治療急性肺損傷和急性呼吸道疾病。其他HNE抑制劑如AZD9668,Ⅱ期臨床試驗結果顯示其pIC50為7.9 nmol/L,Ki為9.4 nmol/L,Kd為9.5 nmol/L,由于脂肪分布異常等嚴重不良反應,現研究已經終止。值得慶幸的是由Bayer公司研發的一種新型具有高選擇性的HNE抑制劑BAY-85-8501具有良好的藥代動力學特性,例如半衰期為8.5 h,口服生物利用度為63%,其Ⅱ期臨床試驗對非囊性纖維化支氣管擴張患者的安全性和有效性進行了評估。另一種口服具有高選擇性的HNE抑制劑MPH996在臨床實驗未達到主要治療終點,目前正在募集受試人群進行Ⅱ期臨床實驗。MMP是一類結構相關的鋅依賴性金屬酶,也是抗炎治療的重要靶點之一。其中,MMP-12對COPD的發生發展起關鍵作用,馬立馬司他(marimastat)是一種非選擇性MMP抑制劑,可顯著抑制MMP-12誘導的早期炎癥,研究者對其進行了腫瘤治療的臨床試驗,但由于關節痛和肌肉骨骼疼痛的副作用限制了其發展。AZ11557272是MMP-9/MMP-12雙靶點抑制劑,豚鼠模型的研究中發現其能減少70%由煙霧引起的總肺活量、剩余容積和肺活量的增加,并防止小氣道重塑[28],有望作為候選藥物進行臨床研究。另一個由AstraZeneca公司研發的MMP-9/MMP-12雙靶點抑制劑AZD1236,已進行了COPD治療的Ⅱ期臨床研究,但目前并沒有報道臨床療效。PR3和HNE是同源蛋白酶,具有調節炎癥反應的特殊功能,但PR3抑制劑的研究正處于起步階段。靶向蛋白酶的藥物臨床研究見表5。

表5 用于COPD的靶向蛋白酶藥物臨床研究

3.2.3 激酶抑制劑 靶向激酶藥物有以下幾類:IKKβ抑制劑、p38 MAPK抑制劑、PI3K抑制劑、JAK抑制劑、EGFR抑制劑。NF-κB通路能誘導多種炎癥基因表達并參與炎性小體的激活和氧化應激過程,IKKβ是NF-κB通路重要的一部分,IMD-3054是一種選擇性IKKβ抑制劑,在氣道炎癥的小鼠模型研究其對NF-κB轉錄活性抑制作用的IC50為1.2μmol/L,前期對該藥前體藥物IMD-1041進行了抗炎測試的Ⅱ期臨床實驗,由于其前藥易產生耐受性的原因導致研究終止。此外,在COPD相關模型中研究了其他IKKβ抑制劑包括BAY65-1942、TPCA-1、PS-1145和ainsliadimer-A的體內外抗炎作用,這些研究為它們作為抗炎藥進行COPD的臨床治療提供了有力的依據[29]。p38 MAPK可以調節多種炎癥基因的表達,口服型p38 MAPK抑制劑是目前研究最為廣泛的一種激酶抑制劑,其中BCT-197、PH-797804、洛吡莫德(losmapimod)和地爾莫德(dilmapimod)等這些藥物已經完成了治療COPD的Ⅱ期臨床試驗。雖然口服型p38 MAPK抑制劑是有效的,但口服大劑量藥物可能會產生全身副作用,因此吸入型p38 MAPK抑制劑的發現是改善治療窗口的另一種策略,目前有部分吸入型藥物正進行治療COPD的臨床評估,其中PF-03715455、AZD-7624、CHF-6297和RV-568已經完成了Ⅱ期臨床試驗,GSK610677進行了Ⅰ期臨床試驗,這些藥物最終能否有效治療COPD還有待進一步研究。PI3Ks能產生脂質第二信使,在免疫系統中起著不可或缺的作用,PI3Kδ和PI3Kγ是PI3Ks的亞型,由GlaxoSmithKline公司研發的GSK2269557是一個吸入型具有選擇性的PI3Kδ抑制劑,在對120名受試者進行為期12周的Ⅱ期臨床試驗的評估中發現其痰中IL-8、IL-6水平降低。TG100-115是一種吸入型具有選擇性的PI3Kδ/γ抑制劑,在哮喘小鼠模型研究中發現TG100-115劑量在1~100μg/kg范圍內可以減少約50%(P<0.05)的氣道高反應[30],并且有良好生物活性、藥代動力學和安全性,可作為候選藥物進行治療COPD的臨床研究。另一種PI3Kδ抑制劑LAS191954在大鼠和犬模型研究中發現其具有良好的PK值,半衰期分別為3.1和10.2 h,具有口服生物利用度高(101%,98%)且清除率低[9.6,1.4 mL/(min·kg)]等特點[31],這些結果顯示,LAS191954有望成為一個口服長效候選藥物。JAK通過磷酸化信號轉導子和轉錄激活子蛋白來調控多種炎癥基因的表達,研究發現吸入型pan-JAK抑制劑PF1367550能減少BEAS-2B細胞和氣道上皮細胞CXCL9,CXCL10,CXCL11的釋放[32],有益于COPD的治療。EGFR可以介導氣道上皮細胞黏液分泌過度和IL-8在氣道上皮細胞中的表達,并且有研究發現EGFR抑制劑AG-1478可以抑制香煙煙霧誘導的體內外黏液素合成[33],由Boehringer Ingelheim公司研發的EGFR抑制劑BIBW 2948為期4周的Ⅱ期臨床試驗結果表明該藥耐受性較差,且有肝功能異常以及FEV1降低等副作用,導致該藥臨床研究被迫終止。靶向激酶的藥物臨床研究進展見表6。

表6 用于COPD的靶向激酶藥物臨床研究
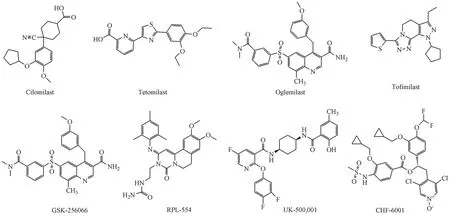
3.2.4 PED4抑制劑 磷酸二酯酶4(phosphodies?terase-4,PDE4)抑制劑具有廣譜抗炎作用,在COPD的治療中有重要意義[34],包括口服型PDE4抑制劑、吸入型PDE4抑制劑、新一代PDE4抑制劑、雙靶點PDE3/4抑制劑。羅氟司特(roflumilast)是唯一批準用于治療COPD的口服型PDE4抑制劑,臨床研究表明,每日劑量500μg可降低COPD的發作頻率并改善肺功能。第二代口服型PDE4抑制劑西洛司特(cilomilast)是治療COPD的首選藥物,Ⅰ期和Ⅱ期臨床研究結果顯示其可顯著改善患者肺功能,但Ⅲ期臨床研究顯示該藥治愈率低,另一個PDE4抑制劑替托司特(tetomilast)具有半衰期長(24 h)、生物利用度高(100%)等特征,目前正進行治療COPD的Ⅱ期臨床試驗。吸入型PDE4抑制劑通過減少全身暴露使副作用最小化和治療窗口最大化,目前許多吸入型PDE4抑制劑正在進行臨床實驗研究,但還沒有藥物被批準使用,其中吸入型PDE4抑制劑UK-500,001由于臨床研究的結果為陰性而被停用,奧米司特(oglemi?last)和妥非司特(tofimilast)正進行研究,其臨床Ⅱ期研究結果尚未更新。由GlaxoSmithKline公司研發的GSK-256066是最有效的PDE4抑制劑(IC50=0.015 nmol/L),對中度COPD患者的Ⅱ期臨床試驗耐受性良好,但其安全性和臨床療效有待進一步驗證[35],另一個吸入型PDE4抑制劑CHF6001(IC50=0.04 nmol/L)的Ⅱ期臨床研究發現其副作用小和治療窗口大,有望進行進一步研究。新一代PDE4抑制劑通過PDE4酶的結構域設計而成的,目前正處于初步研究階段。聯合抑制PDE3和PDE4可能具有附加的抗炎和支氣管擴張作用,近幾年研究發現雙靶點PDE3/4抑制劑對氣道炎性疾病有益,RPL554是一種吸入型雙靶點PDE3/4抑制劑,正在進行臨床Ⅱ期試驗,還有其他4種雙重PDE3/4抑制劑,包括扎達維林(zardaverine),苯扎芬林(benzafentrine),普馬芬群(pumafentrine),托拉芬群(tolafentrine)由于選擇性較差,在臨床實驗出現各種副作用,比如DOA(dead on arrival),指到醫院已死亡,停止對其進行一切醫療行為的病例,而終止臨床研究。PED4抑制劑臨床研究情況如表7。

3.2.5 腺苷受體調節劑 與正常人相比,COPD患者支氣管肺泡細胞外腺苷水平升高,因此腺苷信號成為研究氣道炎性疾病的一個重要靶點。腺苷受體(adenosine receptors,ARs)參與COPD的生理過程且在炎癥細胞和基質細胞廣泛表達。由Pfizer公司研發的A2AAR激動劑UK-432,097在Ⅱ期臨床試驗中評估了對COPD患者的安全性和有效性,現在其研究已終止,另一種A2AAR激動劑GW-328267X在Ⅰ期臨床研究中評估了其安全性、耐受性、藥代動力學和藥效學,其療效還有待進一步研究。目前,這類藥物正處于研究的初步階段,進入臨床研究的藥物還比較少。靶向ARs藥物臨床研究見表8。

表7 用于COPD的PDE4抑制劑臨床研究

3.3 抗氧劑
ROS會破壞內源性蛋白酶/抗蛋白酶的平衡,加速肺損傷,針對抗氧化作用治療COPD的藥物有以下幾類:黏痰溶解藥、Nrf2激活劑、NOX抑制劑、MPO抑制劑、超氧化物歧化酶。N-乙酰半胱氨酸(N-acetylcysteine)、厄多司坦(erdosteine)和羧甲司坦(carbocisteine)等黏液溶解劑具有抗氧化和抗炎特性,在臨床上用于減少COPD患者黏液,有利于氣道引流通暢,改善通氣。轉錄因子Nrf2可以調節多種抗氧化蛋白基因的表達,Nrf2激活劑Sulforaphane在臨床治療中被發掘,但Ⅱ期臨床研究結果顯示其既不影響Nrf2通路,也不影響抗氧化劑或炎癥水平。NOXS是ROS的主要來源[36],目前研究發現的選擇性NOX4抑制劑不多,已知的NOX2和NOX4雙靶點抑制劑有VAS2870,具有抑制超氧陰離子產生(IC50=10.6μmol/L)和抑制氧化低密度脂蛋白誘導的人內皮細胞ROS形成的作用[37],另一種NOX4抑制劑GKT136901具有良好的體外藥代動力學特性,有潛力成為新型抗氧化劑的先導化合物[38]。中性粒細胞和巨噬細胞通過釋放MOP并催化其形成強氧化劑,從而放大氧化損傷和肺部組織炎癥。有研究報道,2-硫黃嘌呤MPO抑制劑AZD5904可以減輕體內由MPO促進的氧化應激,由Pfizer公司研發的MPO抑制劑PF-06282999在Ⅰ期臨床實驗中評估了其在受試者的安全性、耐受性和藥代動力學性質,這些藥物目前還有待進一步的研究。超氧化物歧化酶(superoxide dismutase,SOD)是一種抗氧化劑,通過清除肺部和血液中的ROS來發揮治療作用。抗氧劑臨床研究情況見表9。

表8 用于COPD的靶向ARs的藥物臨床研究


表9 用于治療COPD的抗氧化藥物臨床研究
4 總結與展望
目前用于COPD臨床治療的藥物主要是支氣管擴張劑和抗炎藥物,盡管各大公司已經找到了許多性質優良的候選化合物,有些藥代動力學性質良好、安全性高的藥物已經進行了臨床研究,但是真正批準上市的藥物甚少,有的藥物批準上市后由于其不良反應停止使用。COPD藥物的研究更多地集中于開發新型吸入型藥物,采用軟性給藥策略合理設計適合于吸入給藥的小分子藥物可以減少長期給藥的不良反應,迄今為止,最有效的COPD藥物是通過吸入進行治療,發現優良的口服型藥物治療慢性阻塞性肺病仍然是一個挑戰。總的來說,研發靶向COPD藥物是一個非常具有挑戰性和吸引力的研究領域。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
遼金歷史與考古(2019年0期)2020-01-06 07:45:20
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
電子制作(2018年11期)2018-08-04 03:26:04
汽車工程學報(2017年2期)2017-07-05 08:13:02
國際商務財會(2017年8期)2017-06-21 06:14:14
電子制作(2017年23期)2017-02-02 07:17:19
