成人原發性噬血細胞綜合征合并EB病毒感染1例
2021-04-21 03:09:38黃磊郭淑利肖蓬莉王旖旎王慧睿
內科 2021年1期
黃磊 郭淑利 肖蓬莉 王旖旎 王慧睿
1 鄭州大學附屬洛陽中心醫院血液內科,洛陽市 471000;2 北京友誼醫院血液內科,北京市 100050
【提要】 噬血細胞綜合征(hemophagocytic histiocytosis,HLH)是一組以發熱,肝、脾腫大,全血細胞減少以及骨髓、肝、脾、淋巴結組織出現噬血現象為主要臨床特征的綜合征。HLH分為原發性和繼發性兩種,多數原發性HLH為家族遺傳性基因病,常少兒起病,而成人HLH患者很少考慮為家族性HLH。本文報道了1例成人首診的家族性HLH,并就其發病過程、診療經過、治療預后進行了總結分析,同時通過對患者及其家屬進行基因測序,描繪了家族HLH的UNC13D基因家系圖,這對成人原發性HLH的診治具有重要的參考價值。
噬血細胞綜合征(HLH)又稱噬血細胞性淋巴組織細胞增多癥,是一組以發熱,肝、脾腫大,全血細胞減少以及骨髓、肝、脾、淋巴結組織出現噬血現象為主要臨床特征的綜合征。患者在多種致病因素的作用下,淋巴細胞、單核細胞和吞噬細胞系統異常激活、增殖,分泌大量炎性細胞因子,導致嚴重甚至致命的炎癥反應發生。HLH分為原發性和繼發性兩大類[1],原發性HLH發病率約為1/50 000,70%~80%的患者在1歲以內發病, 90%以上的患者在2歲以內發病,大多患者有家族史。繼發性HLH多由感染(包括細菌、真菌、病毒等,尤其是EB病毒是最主要的誘因)、腫瘤、自身免疫性疾病等誘發,多發生于成年人,患者有明確的原發病,其臨床特征與原發性HLH相似。
1 原發性HLH的類型及診斷
原發性HLH包括:(1)家族性HLH,通常發生在嬰幼兒;(2)免疫缺陷綜合征相關的HLH,如Chediak-Higashi綜合征、Griscelli綜合征、Hermansky-Pudlak綜合征等;(3)EB病毒(EBV)驅動的HLH,如X連鎖淋巴組織增生綜合征(XLP)。其中最常見的是家族性HLH,它是一種常染色體隱性遺傳病分為5個亞型,FHL-1相關缺陷基因及編碼蛋白至今仍未確定;FHL-2至FHL-5則分別對應了PRF1、UNC13D、STX11及STXBP2基因及相關編碼蛋白[2]。依據HLH-2004診斷標準[3],經分子生物學檢查明確存在家族性,或已知有遺傳缺陷(包括PRF1、UNC13D、STX11、STXBP2、Rab27a、LYST、SH2D1A、BIRC4、ITK等基因病理性突變),或符合以下8項指標中的5項即可診斷為HLH。(1)持續發熱時間>7 d,體溫>38.5℃;(2)脾臟腫大(肋緣下≥3 cm);(3)外周血血細胞減少(累及兩系或三系):血紅蛋白(HGB)<90 g/L,血小板(PLT)<100×109/L、中性粒細胞<1.0×109/L且非骨髓造血功能減低所致;(4)三酰甘油水平升高和(或)纖維蛋白原水平降低:三酰甘油>3 mmol/L或高于同年齡參考值的3個標準差,纖維蛋白原<1.5 g/L或低于同年齡參考值的3個標準差;(5)在骨髓、脾臟或淋巴結內找到噬血細胞;(6)自然殺傷(NK)細胞活性降低或缺如;(7)血清鐵蛋白≥500 μg/L;(8)可溶性IL-2受體(sIL-2R或sCD25)水平升高。
2 病例資料
患者男,42歲,于2019年2月20日因“間斷發熱1周”入院。入院前1周,患者勞累后發熱,體溫最高39℃,伴畏寒,頭痛、頭暈,咳嗽、咳白痰,雙下肢肌肉疼痛,全身乏力,到我院門診查血常規示:白細胞5.88×109/L、中性粒細胞39.6%、淋巴細胞50.2%、血紅蛋白136 g/L、血小板88×109/L;CRP正常,甲/乙型流感監測結果陰性。門診行抗病毒治療效果欠佳,遂住院治療。患者2年前因“三系減少、脾大”于外院給予脾臟切除術(病理示:淤血脾)治療,之后彩超檢查發現肝臟增大,但未再進行進一步診治。
入院后復查血常規示白細胞3.67×109/L、中性粒細胞39.6%、淋巴細胞50.2%、血紅蛋白108 g/L、血小板63×109/L;實驗室檢查發現LDH 258 U/L、FER 600 ng/mL;EB病毒抗體陽性,多次檢查顯示EBV-DNA明顯升高;腦脊液檢查提示隱球菌感染。腹部CT示肝腫大、肺部感染、胸腔積液、心包積液、腹膜后多發淋巴結腫大。骨髓象示增生活躍,異型淋巴細胞占9%~20%,細胞大小不等,呈不規則形、梭形等。流式免疫表型檢測,可見一群表型為CD34-,CD117-,CD2+,cCD3+,CD20-,CD19-,CD5-,CD10-,mCD3-,CD4-,CD8-,CD7-,CD56-,CD16-,CD94-,CD30-細胞;CD45強表達,占有核細胞的9.1%,胞體較大,為異常T淋巴細胞。骨髓活檢提示骨髓增生極度活躍、淋巴細胞比例增高;EBV+淋巴細胞增殖性疾病;慢性活動性EB病毒感染(CAEBV)不除外。TCR重排(T細胞受體基因重排)陽性;全身PET-CT掃描陰性。淋巴結活檢示淋巴細胞反應性增生;EB病毒原位雜交(+)[2年前脾臟活檢標本重新閱片示:竇內大量淋巴組織浸潤,并有噬血現象;EB病毒原位雜交散在(+)]。sCD25升高為2 777 U/mL,NK細胞活性減低為3.57%。患者合并EBV感染,根據HLH-2004診斷標準,診斷為EB病毒感染繼發的HLH。給予莫西沙星、頭孢唑肟抗感染治療,給予更昔洛韋、帕拉米韋抗病毒治療,給予兩性霉素B、氟胞嘧啶聯合氟康唑抗隱球菌治療,采用VP-16聯合地塞米松化療方案規范治療。
專家會診討論后,考慮到該患者病史較長、長期處于免疫缺陷狀態、合并隱球菌感染、多系統受累,擬診斷為隱匿起病的原發性HLH。為此,對該患者進行了HLH相關致病基因套組二代測序,篩查噬血相關基因外顯子編碼區域突變,結果發現:患者存在UNC13D基因部分位點突變,包括UNC13D基因Exon10上存在無義突變c.766C>T(p.Arg256Ter)(雜合,變異頻率51.3%),可能為致病位點(見圖1);UNC13D基因Exon6上存在錯義突變c.518C>T(p.Thr173Met)(雜合,變異頻率50.33%,PolyPhen-2 humVAR值為0.991,SIFT為0.00),臨床意義暫不明確(見圖2)。據此,患者被確診為原發性HLH-家族性HLH。經抗感染、抗隱球菌治療及采用VP-16聯合地塞米松化療方案控制噬血癥狀后,患者發熱、肝臟腫大等癥狀逐漸好轉出院,目前接受門診治療、隨訪,患者口服環孢素維持,血常規基本穩定,LDH、鐵蛋白逐漸降低至正常;患者與其大哥配型提示半相合,下一步擬行半相合造血干細胞移植治療。

圖1 患者UNC13D 基因Exon10測序結果
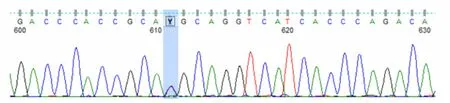
圖2 患者UNC13D 基因Exon6測序結果
3 患者的原發性HLH家族史
患者被確診后,對其家系成員進行了UNC13D基因外顯子編碼突變篩查發現,可能致病位點來自其母親(UNC13D基因Exon10上無義突變c.766C>T)(見圖3),其二哥[UNC13D基因Exon10上錯義變異 c.766C>T(p.Arg256Ter)](見圖4)和兒子[UNC13D基因 Exon10上存在無義變異 c.766C>T(p.Arg256Ter)](見圖5)也存在該位點突變,但3人均無相關病史。患者父親未被證實致病的基因變異[UNC13D基因 Exon6上存在錯義變異 c.518C>T(p.Thr173Met)](見圖6);患者大哥無明顯致病基因突變(見圖7)。檢查結果進一步佐證了患者為家族性HLH(見圖8)。
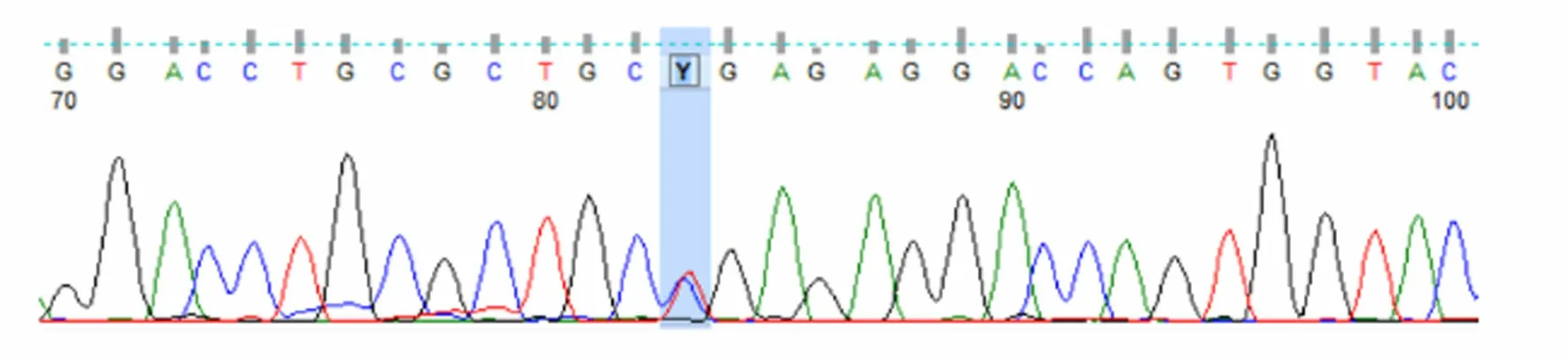
圖3 患者母親UNC13D基因Exon10測序結果
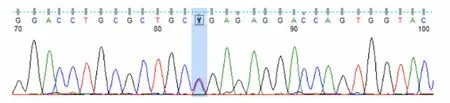
圖4 患者二哥UNC13D基因Exon10測序結果
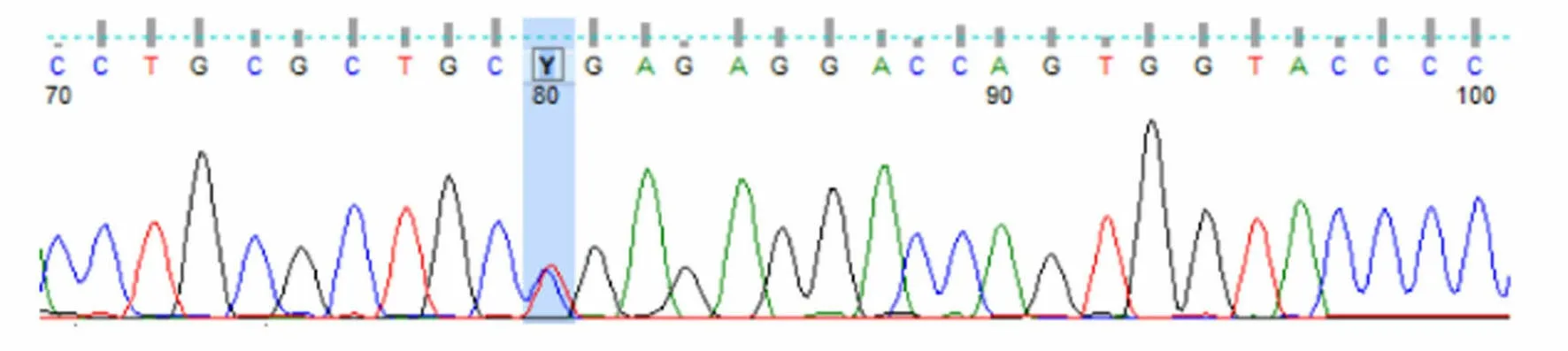
圖5 患者兒子UNC13D基因Exon10測序結果
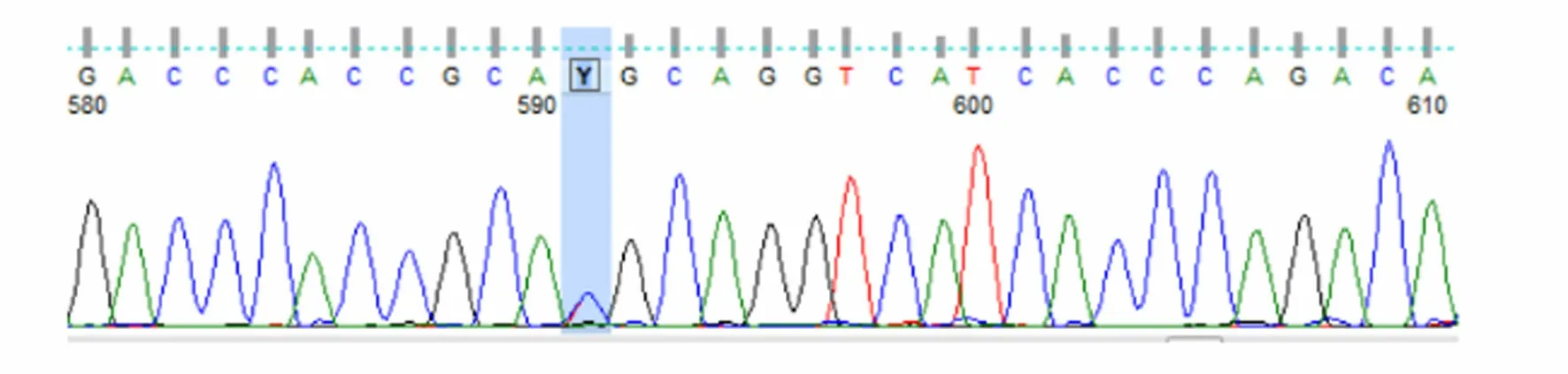
圖6 患者父親UNC13D 基因 Exon6測序結果

圖7 患者大哥UNC13D基因外顯子測序結果正常
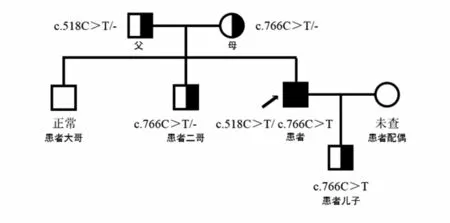
圖8 患者家族性HLH的UNC13D基因家系圖
4 討 論
原發性HLH患者多為遺傳性HLH,現有報道的患者幾乎均為嬰幼兒,成人HLH常被診斷為繼發性HLH。近年來,隨著基因診斷技術的發展,陸續5個家族性HLH相關基因(UNC13D、PRF1、STX11、SH2D1A、RAB27A)突變被鑒定和認可,國際組織細胞協會據此制定了HLH-2004診斷指南,明確把基因缺陷作為診斷和鑒別原發性家族性HLH的標準。Wang等[4-6]相繼通過HLH相關基因突變檢測確診了多例成人家族性HLH患者,其中Sieni等報道的11例成人家族性HLH患者的年齡為18~43歲,中位年齡為23歲。
本例患者反復發熱、全血細胞減少、肝脾淋巴結腫大、血清鐵蛋白水平升高、血清EBV-DNA陽性、合并中樞神經系統隱球菌感染、NK細胞活性明顯下降、可溶性CD25升高,根據HLH-2004診斷標準,開始時初步考慮為EB病毒感染繼發的HLH。在抗感染、抗隱球菌治療及化療后,患者發熱、肝臟腫大等癥狀逐漸好轉,血清EBV-DNA轉陰,腦脊液檢測結果恢復正常;患者病史較長、長期處于免疫缺陷狀態并合并隱球菌感染,不排除患者為原發性HLH的可能,故對患者進行HLH相關基因二代測序,結果證實患者存在UNC13D基因突變,最終診斷為原發性HLH合并EB病毒感染。通過對患者家系成員行HLH相關基因測序發現,患者的可能致病位點來自其母親UNC13D基因 Exon10(存在無義突變),其二哥和兒子也同時存在該位點突變,其父親在UNC13D基因Exon6上存在錯義突變但臨床意義暫不明確。據此,最終確診該患者為家族性HLH。由此可見,對成人HLH患者進行HLH相關基因篩查十分必要。
HLH患者預后差,病死率較高,Rivière 等[7]報道,成人HLH患者的病死率達58%。目前HLH患者的一線治療方法有HLH-94方案和HLH-04方案,HLH-04方案較HLH-94方案增加了環孢素A(cyclosporine A,CsA)。CsA可降低T細胞活性,減少炎性因子分泌,但CsA肝腎毒性明顯。本例患者病情較重,發病時合并神經系統隱球菌、EB病毒感染,CsA應用受限,考慮到HLH-04治療方案不具有優勢[8],因此選用HLH-94方案進行治療。患者經化療、抗感染治療后,病情好轉,目前門診隨診,口服CsA維持治療;患者體溫正常、肝臟體積和肝功能、LDH、鐵蛋白正常,病情穩定。由此可見,對確診的成人家族性HLH患者積極進行免疫治療及化療,同時加強抗感染、對癥支持治療,能較好地控制病情發展。
化療可逆轉HLH患者的超炎癥狀態,造血干細胞移植治療可恢復HLH患者對病原體正常的免疫反應,移植后5年生存率可達71%,5年無事件生存率可達60%[9]。移植的最佳供者為不攜帶HLH相關基因全相合同胞,其次為非血緣供者,不攜帶基因單倍體者亦可[10]。本例患者被確診為家族性HLH,目前雖病情穩定,但病情可能會隨時惡化而危及生命,因此即使只有半相合供者,也強烈推薦其進行移植治療。在誘導緩解的基礎上進行移植治療,可有效提高移植的成功率和患者的長期存活率[11]。移植治療原發性HLH的移植相關死亡率高達30%,常因感染、肝靜脈閉塞癥及非感染性肺炎所致,移植排斥率也較高[12]。本患者大哥未檢出噬血相關基因突變,且配型為半相合,故優選其大哥為供者捐獻造血干細胞。考慮患者穩定期可能極短,因此積極動員患者盡快行同胞半相合造血干細胞移植治療,目前正處于準備階段。
HLH發展迅速,病死率高,臨床上對反復發熱、肝、脾、淋巴結腫大,全血細胞減少的患者需高度警惕,盡快完善鐵蛋白、血脂、NK細胞功能、CD107a等相關檢查,明確診斷。對成人HLH仍不能排除家族性HLH的可能,對病因不明確、病史較長或反復發作的HLH患者,需盡快完善基因測序排除家族性HLH可能;對繼發性HLH患者可針對病因進行治療。
