三氯乙酰亞胺酯法合成紫草素-1′-O-β-D-吡喃葡萄糖苷
2021-04-20 00:29:34李龍珠張玉心吳成柱李紅梅
蚌埠醫學院學報 2021年3期
李龍珠,張玉心,吳成柱,李紅梅,馬 濤
紫草素是從我國傳統中藥紫草中分離得到的一種脂溶性萘醌類化合物[1],是紫草的主要活性成分。研究表明紫草素具有抗腫瘤[2-4]、抗炎[5]、抗病毒[6]、抗菌[7]、促進傷口愈合[8]等藥理作用。與其他萘醌類化合物類似,紫草素的穩定性較差,且在水中的溶解度低,這在一定程度上阻礙了紫草素的開發與應用[9]。
糖是自然界中存在最為廣泛的生物分子,幾乎存在于所有生命體中,通過糖苷化修飾可以提高化合物的水溶性和穩定性[10-11]。本課題組前期采用酶催化法已經制得兩種紫草素吡喃葡萄糖苷,其穩定性均顯著高于紫草素,水溶性相較紫草素增加上千倍,兩種紫草素吡喃葡萄糖苷在體外抗腫瘤作用相較紫草素有所下降[12],但在后續開展的肝癌裸鼠移植瘤預實驗中,紫草素-1′-O-β-D-吡喃葡萄糖苷的抑腫瘤作用明顯優于紫草素。為了對其體內抗腫瘤作用進行全面研究,需要大量制備紫草素-1′-O-β-D-吡喃葡萄糖苷用于裸鼠移植瘤實驗及藥代動力學研究。但是在實驗室條件下,通過酶催化法所得糖苷化合物的量只有微克級,難以滿足動物實驗的需要,而且酶的制備需要經過菌群培養、基因表達、構建重組質粒、提純等多個步驟,這也導致了酶的制備成本高[13],因此迫切需要建立一種成本低、操作簡便且適于大量制備紫草素-1′-O-β-D-吡喃葡萄糖苷的方法。
目前已有研究[14]采用三氯乙酰亞胺酯法將多種乙酰基取代的糖基加在紫草素1′-OH上,制得了11種紫草素苷,尚未見化學法合成紫草素-1′-O-β-D-吡喃葡萄糖苷的報道。本研究選擇β-D-吡喃葡萄糖為起始原料,經選擇性乙酰化后,再進一步制得糖基三氯乙酰亞胺酯作為糖苷供體,選擇催化效能更佳的三氟甲磺酸三甲基硅脂來催化紫草素糖基化反應,制得紫草素-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷,利用甲醇鈉/甲醇醇解脫乙酰基后制得紫草素-1′-O-β-D-吡喃葡萄糖苷,并采用核磁共振法、質譜法對各產物的結構進行了鑒定。
1 材料與方法
1.1 儀器與試藥 IKA RV10型旋轉蒸發儀(德國 IKA 公司);LC-20A型高效液相色譜儀(日本島津公司);B23-2型恒溫磁力攪拌器(上海碩光司樂儀器有限公司);梅特勒-托利多XP250型天平(瑞士梅特勒-托利多公司);BRUKER ASCENDTM400 MHz核磁共振光譜儀(美國Bruker公司);Thermo TSQ quantis液質聯用儀(美國Thermo Fisher Scientific公司)。
甲醇、乙酸乙酯、石油醚、乙醇、無水乙醚(上海泰坦化學有限公司)均為分析純;左旋紫草素標準品(中國藥品生物制品檢定所,批號110769-200506);乙酸酐(江蘇彤晟化學試劑有限公司);4A分子篩、吡啶(薩恩化學技術有限公司);β-D-葡萄糖、四氫呋喃、1,8-二氮雙環(5,4,0)-7-十一烯、三氯乙腈、乙酸肼、碳酸氫鈉(上海麥克林生化科技有限公司);三氟甲磺酸三甲基硅脂(阿達瑪斯試劑有限公司)。
1.2 方法 以二氯甲烷為溶劑,三氟甲磺酸三甲基硅脂為催化劑,在嚴格無水和N2保護下,通過自制的2,3,4,6-四-O-乙酰基-β-D吡喃糖酰基2,2,2-三氯乙酰亞胺酯與紫草素發生糖苷化反應,有效合成了紫草素-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷,并在甲醇鈉/甲醇體系中將乙酰基醇解為羥基,最終得到目的產物紫草素-1′-O-β-D-吡喃葡萄糖苷(見圖1)。
1.2.1 1,2,3,4,6-五-O-乙酰基葡萄糖(1)的合成 取乙酸酐56.7 mL和吡啶5 mL混勻后分多次加入至18 gβ-D-葡萄糖中,冰浴條件下攪拌,并緩慢升至室溫反應過夜。反應結束加入25 mL蒸餾水攪拌,再用25 mL乙酸乙酯萃取2~3次,取有機相用10%飽和碳酸氫鈉水溶液中和反應產生的副產物醋酸及過量的乙酸酐,再用水洗3次,每次20 mL,上層有機層用旋轉蒸發儀除去溶劑,得到的粗產物于室溫下用75 mL乙醇結晶,得到白色粉末,采用核磁共振波譜(nuclear magnetic resonance spectroscopy,NMR)和電噴霧-質譜(electrospray ionization mass spectrometry,ESI-MS)表征其結構。
1.2.2 2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖(2)的合成 將20 g化合物(1)和5.2 g乙酸肼加入到10 mL四氫呋喃中溶解,室溫下反應4 h,旋轉蒸發除去溶劑,用10 mL二氯甲烷溶解殘余物,將有機相用水洗3次,每次5 mL,旋轉蒸發除去溶劑得粗產物,加入20 mL無水乙醚和20 mL石油醚后于冰水浴下析出結晶,得到白色粉末,采用NMR和ESI-MS表征其結構。
1.2.3 2,3,4,6-四-O-乙酰基-β-D吡喃糖酰基2,2,2-三氯乙酰亞胺酯(3)的合成 取9 g化合物(2)和10 mL三氯乙腈依次加入至有50 mL無水二氯甲烷的燒瓶中,冰浴條件下攪拌30 min,再緩慢滴加1.5 mL的1,8-二氮雙環(5,4,0)-7-十一烯,撤除冰浴,在N2保護下室溫反應過夜。旋轉蒸發溶劑后得黃褐色油狀物,油狀物經硅膠柱層析(石油醚∶乙酸乙酯=8∶1)純化,最終得白色粉末,采用NMR和ESI-MS表征其結構。
1.2.4 紫草素-2,3,4,6-四-O-乙酰基-β-D-吡喃葡萄糖苷(4)的合成 將10 g紫草素和18.8 g化合物(3)溶解于70 mL無水二氯甲烷中,加入4A分子篩粉末,N2保護下室溫攪拌30 min,然后緩慢滴加0.63 mL三氟甲磺酸三甲基硅脂,于-20 ℃反應8 h。抽濾除去分子篩,將有機相用水洗3次,每次50 mL,旋轉蒸發除去溶劑后,將粗產物用乙酸乙酯復溶,經硅膠柱層析(石油醚∶乙酸乙酯=5∶1)純化,最終得紅色粉末,采用NMR和ESI-MS表征其結構。
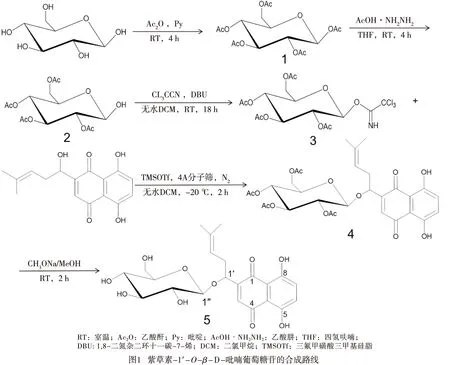
1.2.5 紫草素-1′-O-β-D-吡喃葡萄糖苷(5)的合成 取10 g化合物(4)溶解于50 mL甲醇中,向其中緩慢滴加甲醇鈉溶液,調節溶液pH至8~9,室溫回流反應2 h,加入冰醋酸調pH為中性并終止反應。旋轉蒸發溶劑后,將粗產物用二氯甲烷復溶后經硅膠柱層析(二氯甲烷∶甲醇=10∶1)純化,最終得紅色粉末,采用NMR和ESI-MS表征其結構。
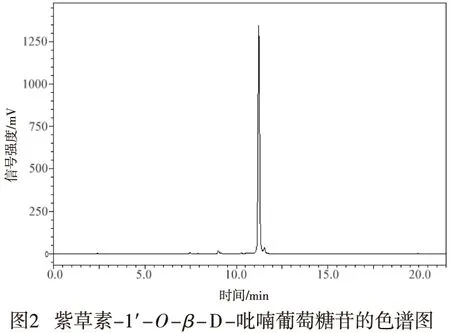
1.2.6 化合物5純度測定 取適量化合物5,加入適量甲醇配制成溶液,取20 μL化合物5的甲醇溶液進高效液相色譜儀分析,以峰面積歸一化法測定化合物5純度。采用SunFireTMprep C18(10 mm×250 mm) 色譜柱,以水-乙腈作為流動相進行梯度洗脫(0~20 min,20%乙腈→100%乙腈),流速為1 mL/min,檢測波長為254 nm。
2 結果
化合物1為白色粉末,得到28.5 g,收率73.1%。1H NMR (400 MHz,CDCl3)δ:5.73 (d,J=8.0 Hz,1H),5.27 (t,J=9.4 Hz,1H),5.15 (m,2H),4.30 (dd,J=4.6,12.5 Hz,1H),4.13 (dd,J=2.2,12.5 Hz,1H),3.86 (m,1H),2.13 (s,3H),2.10 (s,3H),2.05 (s,6H),2.03 (s,3H)。13C NMR(100 MHz,CDCl3)δ:170.6,170.1,169.4,169.2,168.9,91.7,72.8,72.7,70.2,67.8,61.5,20.8,20.7,20.6,20.6,20.5。ESI-MS(m/z):413.15[M+Na]+。
化合物2為白色粉末,得到12.8 g,收率71.7%。1H NMR (400 MHz,CDCl3)δ:5.27 (t,J=9.5 Hz,1H),5.10 (t,J=9.9 Hz,1H),4.90 (dd,J=8.0,9.7 Hz,1H),4.76 (t,J=8.6 Hz,1H),4.25 (m,2H),3.77 (m,1H),3.66 (d,J=8.8,1H),2.11 (d,J=1.4,6H),2.04 (d,J=3.8,6H)。13C NMR(100 MHz,CDCl3)δ:170.9,170.7,170.1,169.6,169.5,95.6,73.3,72.2,68.5,62.0,20.7,20.7,20.6,20.6。ESI-MS(m/z):371.13[M+Na]+。
化合物3為白色粉末,得到12.4 g,收率57.8%。1H NMR (400 MHz,CDCl3)δ:8.69 (s,1H),6.63 (d,J=4.0 Hz,1H),5.58 (dd,J=1.2,3.1 Hz,1H),5.45 (dd,J=3.1,10.8,1H),5.39 (dd,J=3.5,10.8 Hz,1H),4.46 (t,J=6.6 Hz,1H),4.15(m,2H),2.19 (s,3H),2.05 (s,3H),2.03 (s,3H),2.04(d,J=2.1,6H)。13C NMR (100 MHz,CDCl3)δ:170.3,170.1,170.1,169.9,161.0,93.6,90.8,69.0,67.5,67.4,66.9,61.2,20.7,20.6,20.6,20.5。ESI-MS(m/z):514.08[M+Na]+。
化合物4為紅色粉末,得到8.7 g,收率68.3%。1H NMR (400 MHz,CDCl3)δ:12.48(s,1H),12.42 (s,1H),7.14 (m,3H),5.15 (m,1H),5.03 (m,3H),4.89 (m,1H),4.58 (d,J=8.1 Hz,1H),4.01 (m,2H),3.57 (m,1H),2.4(m,1H),2.24 (m,1H),2.12 (s,3H),1.98 (s,3H),1.95 (s,3H),1.87(s,3H),1.60 (s,3H),1.47 (s,3H)。13C NMR (100 MHz,CDCl3)δ:181.2,178.6,170.2,170.0,169.1,168.5,165.1,164.8,149.2,135.0,134.1,13.7,131.5,119.2,112.1,111.6,101.7,75.5,73.2,72.0,71.4,69.1,61.9,33.5,25.8,20.8,20.6,20.4,20.3,17.8。ESI-MS(m/z):641.26[M+Na]+。
化合物5為紅色粉末,得到4.9 g,收率67.3%。1H NMR (400 MHz,CDCl3)δ:7.28(s,3H),7.21(s,3H),5.37(t,J=14.8,1H),5.14(br s,1H),4.64(d,J=7.7,1H),3.23(m,1H),3.26(m,2H),3.27(m,2H),3.53(m,1H),3.72(m,1H),2.63(s,1H),2.62(m,1H),2.64(m,1H),2.53(m,1H),1.66(s,3H),1.52(s,3H)。13C NMR (100 MHz,CDCl3)δ:183.5,183.3,175.2,174.4,151.7,136.8,135.6,135.0,133.8,120.7,115.6,113.5,104.7,79.6,78.4,75.2,73.5,71.8,63.8,35.5,26.7,18.8。ESI-MS(m/z):473.19[M+Na]+。
根據ESI-MS和NMR結果分析其分子式為C22H26O10,其共振譜與紫草素相似,除了來自葡萄糖基部分的額外核磁信號,基于葡萄糖異頭位質子的耦合常數(為7.7 Hz),包括從δH 4.64(d,J=7.7,1H,H-1″)到δC 75.2(C-1′)的異核多鍵關聯表明葡糖基部分已經連接到紫草素C-1′處,化合物5的結構被鑒定為紫草素-1′-O-β-D-吡喃葡萄糖苷。總收率為38.9%,峰面積歸一化法測定其純度為96.7%,色譜圖見圖2。
3 討論
相較于酶催化法,化學合成法制備糖苷類化合物具有原料易得,成本低的優勢。常用的化學糖基化方法主要有Koenig-Knorr法[15]、三氯乙酰亞胺酸酯法[16]和相轉移催化法[17]等。Koenig-Knorr法產率低、區域選擇性差、反應后處理繁瑣,并且使用價格昂貴且環境污染較大的汞、銀鹽作催化劑,近年來該方法的使用在逐漸減少。相轉移催化法具有反應速度快,反應條件溫和,后處理簡單的特點,但是Koenig-Knorr法和相轉移催化法在合成糖基供體時常以苯乙酰基或乙酰基保護的溴代糖作中間體,溴代糖對制備和貯藏的條件要求十分苛刻,且極易分解,會給下一步的糖基化反應帶來很大影響。
三氯乙酰亞胺酯法制備糖苷類化合物不需要重金屬催化劑,多采用糖基三氯乙酰亞胺酯作為糖苷供體,與其他糖基給體相比,糖基三氯乙酰亞胺酯更適合于氧苷類化合物的合成且不易形成原酸酯副產物,由于有鄰基參與效應,糖基三氯乙酰亞胺酯法具有高立體選擇性和高收率的優點[18],近年來在寡糖和糖苷的合成中得到了廣泛的應用,逐漸成為首選方法[13]。
三氯乙酰亞胺酯法對底物的要求較高,反應體系要嚴格除水,因此在合成化合物4時采用加入分子篩的方法以除去反應體系中的水,經過預試驗探索,最終選擇了除水效果最好的4A分子篩。在前期實驗摸索時,采用了文獻[14]中的三氟化硼乙醚溶液作為糖基化反應催化劑,反應得到較多副產物,目標產物產率低,不易純化,后將催化劑更換為三氟甲磺酸三甲基硅脂,化合物4的得率顯著提高,收率達到57.8%,且副產物減少。考慮到糖苷鍵易斷裂,在化合物4除去乙酰基制備化合物5時不可采用水解的方式,本研究選擇無水甲醇為溶劑,采用甲醇鈉/甲醇體系對化合物進行醇解,且反應需要在低溫、無水的條件下進行,達到減少脫水物雜質,提高產物收率的目的。
三氯乙酰亞胺酯法用于紫草素-1′-O-β-D-吡喃葡萄糖苷合成,具有反應溫和、操作簡單、高立體選擇性的優點,也為其他天然物質的糖基化修飾提供了參考。
