可見光催化炔烴的含氟雙官能團化的研究進展
2021-04-14 04:58:26邱江凱
生物加工過程 2021年2期
袁 鑫,邱江凱,郭 凱
(南京工業大學 生物與制藥工程學院,江蘇 南京 211800)
炔烴為一類含有不飽和碳—碳三鍵的化合物,通過碳原子sp雜化,其獨特的結構使分子具有較高的反應活性[1]。如何利用炔烴構建有價值的分子骨架一直廣受人們的關注。其中,炔烴的雙官能化是一種合成簡單功能化化合物的有效方法,可以將炔烴轉化為多種有機骨架。
太陽光是清潔的可再生能源,是大自然贈予人們的寶貴財富。1984年,Canoy等[2]首次報道了以[Ru(bpy)3](BF4)2作為光催化劑,在藍光LED燈的照射下,以較高收率實現了Pschorr反應。然而在可見光區域內,可見光的能量不能直接引發常見的有機小分子化合物,必須依靠可見光的光敏劑引發,促使電子轉移,從而實現光反應。因此,在之后的20年內,光催化反應并沒有得到有機化學家們的關注,直至金屬多吡啶絡合物和有機染料可以在很溫和的條件下,作為光敏劑參與反應被廣泛報道。2008年,Ischay等[3]報道了以[Ru(bpy)3]Cl2作為光敏劑,在可見光光催化條件下,實現了分子內[2+2]環的加成反應。同年,Nicewicz等[4]報道了以[Ru(bpy)3]Cl2為光催化劑,首次將可見光催化劑和有機小分子催化結合,從而實現了α-位不對稱烷基化的反應。這些具有重要意義的工作使光催化重新進入有機化學家們的視野,并得到了長遠的發展。近十年來,可見光催化在反應方法學上有了突飛猛進的發展。
在光催化循環中,光敏劑吸收可見光產生活性中間體,這些激發態中間體具有較強的氧化和還原能力,通過電子轉移的方式將光能轉化為化學能。光循環機制分為兩類,分別是氧化淬滅和還原淬滅循環。在有機化合物氧化劑和還原劑的存在下,激發態中間體被電子供體還原淬滅,而電子受體淬滅則為氧化淬滅。相比不穩定的自由基引發劑,光敏劑并不直接作為金屬氧化還原劑參與反應,而是在溫和的條件下,傳遞來自有機化合物氧化劑和還原劑的電子,其安全高效的特點符合綠色化學的理念。而光氧化還原催化和金屬雙循環催化的機制途徑一般分為氧化還原途徑或能量轉移途徑[5]。
在還原性淬滅途徑中,激發態光催化劑可以被電子給體還原,從而得到作為強還原劑的物種。相應的,激發態光催化劑可以被電子受體氧化以提供PCox物種。除了光誘導分子內電荷轉移,通過非共價相互作用組裝的給體和受體之間的光誘導的分子間電荷轉移,也是光化學界中眾所周知的反應過程。
近十年來,在現代光致氧化還原催化領域中,經過國內外學者的引領,光催化反應歷經了光致氧化還原-小分子雙重催化[6]、光致氧化劑-過渡金屬雙重催化[7]、光致氧化劑-有機小分子-有機小分子三重催化[8]、光致氧化劑-有機小分子-過渡金屬三重催化[9]、光致氧化劑-酶催化[10]和三苯基膦-碘化鈉體系催化[11]的發展歷程,且每一次新的發現都預示著一個全新的研究方向。
近年來,可見光催化炔烴的氟烷基化、氧化偶聯、烯烴和炔烴的雙官能團化領域均取得了重要進展。含氟類化合物由于其溶解度和親酯性的變化,比非含氟類似化合物具有更好的膜滲透性和更高的生物利用度。炔烴氟化最直接有效的手段通常為自由基試劑、親核試劑或者親電試劑對炔烴進行的氟化雙官能團化。筆者將基于近年來不同類型的氟化試劑對可見光催化炔烴的含氟雙官能團化的研究進展進行總結。
1 自由基三氟甲基試劑
1.1 全氟碘代烷烴作為氟源
全氟碘代烷烴(通式CF3(CF2)nI)在合成藥物分子、農藥和材料等分子結構中一直占據著重要地位,在過去的幾十年里受到了人們廣泛的關注[12]。傳統的方法是在短波紫外線(10~400 nm)輻射下,使用全氟烷基鹵代物(Rf-X)對烯烴進行自由基全氟烷基化,但該方法通常存在選擇性差、收率低以及反應條件苛刻等問題[13]。Haszeldine[14]早在1949年就報道了在光照或者加熱的情況下,三氟甲基碘甲烷(CF3I)會激發產生三氟甲基自由基,在乙烯存在的情況下,三氟甲基自由基會加成到烯烴末端,以生成相應的自由基中間體,該自由基中間體從三氟甲基碘甲烷中捕獲一個碘原子,進而產生3-碘-1,1,1-三氟丙烷和三氟甲基自由基(圖1)。
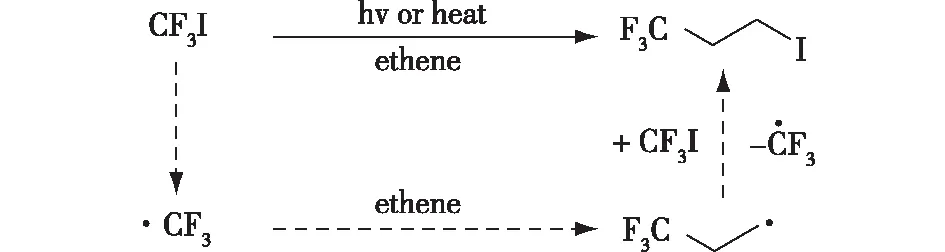
圖1 CF3I生成·CF3的歷程[14]Fig.1 The generation of ·CF3 radical from CF3I[14]
該工作被報道后,三氟甲基碘甲烷作為碳自由基前體的這種分子間電子轉移/自由基加成(ATRA)已經成功被應用于各種炔烴。Me3Al[15],Et3B/O2[16],Na2S2O4[17],ET2ZN[18],FeSO4/H2O2/DMSO[19]等體系已經被廣泛應用于引發三氟甲基自由基對炔烴的加成。近年來,由于可見光(400~760 nm)照射下光氧化還原催化體系的發展,自由基全氟烷烴化都得到了長遠的發展[20-21]。
2012年,Wallentin等[22]提出了一種非常有效的碘代氟代烷基化的方法,利用釕或者銥的在藍光(375~450 nm)照射下的聯吡啶配體絡合物的光氧化還原催化劑,將全氟碘代烷烴引入烯烴和炔烴中進行氟化反應,該方法條件溫和,可以在0.5 h反應得到最高可達99%產率的目標產物。在此研究中,作者提出原子轉移自由基加成ATRA的反應機制,實驗結果表明,[Ir{(CF3)ppy}2(dtbbpy)]PF6的氧化淬滅可以有效應用于ATRA。另外,反應條件Ru(bpy)3Cl2的還原淬滅循環和抗壞血酸鈉在可見光照射下作為犧牲電子供體,全氟烷基碘通過原子轉移自由基加成ATRA的途徑對炔烴可進行有效的加成反應。這些結果表明:光催化劑的氧化和還原淬滅途徑可以相互互補,通過修改反應條件可以控制反應構型(圖2)。該方案雖然可以有效介導末端炔烴與活化的烷基溴化物和碘化物之間的ATRA,但是反應條件不適合活化的烷基氯,1,2-二取代的烯烴或苯乙炔衍生物。

圖2 原子轉移自由基加成的機制路徑[22]Fig.2 The mechanism of ATRA [22]
2014年,Iqbal等[23]發表了類似的工作,即采用同樣的原始底物,利用Ru(phen)2+催化劑作為引發劑,通過控制催化劑、溶劑以及堿等細微的差異,經過ATRA的機制過程,獲得炔烴的不同產物,如:碘三氟甲基化,氫三氟甲基化和三氟甲基化 (圖3),但是該部分工作并不能很好地控制加成產物的構型。2016年,該課題組又成功地將Ru(phen)3+應用到該催化體系中[24]。值得注意的是,Choi等[25]采用無機電子鹽[Ca2N]+·e-作為電子源的自由基介導反應,對未活化的炔烴進行氫氟甲基化,在轉化過程中,陰離子從[Ca2N]+·e-的電子轉移到三氟甲基化試劑全氟碘代烷烴,以引發自由基介導的三氟甲基化,反應中使用的乙腈同時充當了溶劑和電子釋放促進劑以及氫原子源。

圖3 不同的反應路徑[23]Fig.3 Different reaction conditions[23]
盡管釕和銥與聯吡啶配體絡合的光氧化還原催化劑具備高效的反應性能,但成本十分昂貴。因此,近年來,人們開始研究光穩定、無毒性以及廉價的有機染料,如:曙紅或者亞甲基藍等在可見光輻射下作為有機光氧化還原催化劑的反應[26]。2017年,Matsuzaki等[27]探索了有機染料曙紅Y作為光催化劑催化炔烴的ATRA反應[26]。除常規有機染料外,人工有機染料也備受關注,Kohei等[28]一直致力于對人工藍色有機染料酞菁的結構進行設計和改造,使溶解性極差的酞菁可以高度溶解于各種有機溶劑中,并將其應用于光催化反應中,比較有代表性的工作為三氟乙氧基涂覆的硼亞酞菁可以在能量較低的紅光(600~700 nm)輻照下催化烯烴和炔烴自由基烷基化,該方法高效溫和,但是隨著反應時間的增長,催化劑的催化活性會隨之消失[27]。隨后,該課題組又發表了在可見光照射下,由三氟乙氧基涂覆的鋅酞菁鋅催化的,包括炔烴在內的烯烴自由基全氟烷基化,成功解決了催化劑體系不穩定的缺點,該反應同樣是經過一個ATRA的機制過程[28]。
為了降低成本,除了發展廉價的有機染料外,另外一種有效的策略也被人們發現,即采用廉價的金屬銅等代替昂貴的銥、釕等金屬與聯吡啶進行配位后,作為光催化劑參與反應。2018年,Rawner等[29]使用菲咯啉銅催化劑代替傳統的多吡啶絡合物金屬有機催化劑,進行光氧化還原催化苯乙烯和苯基乙炔的碘全氟烷基化反應。與常用的[Ru(bpy)3]Cl2,[Ru(phen)3]Cl2或fac-Ir(ppy)3相比,[Cu(dap)2]Cl能夠更加有效地將苯乙炔轉化為相應的全氟烷基,同位素標記的乙苯指出銅催化劑除了光致電子轉移外,還具有其他作用。在此工作中,還首次提出了涉及Cu(III)中間體或[CuI]+中間體的配體的分子內催化循環。
隨后,人們將注意力轉向對炔烴底物的結構改造,即將烯烴與炔烴相連,形成1,n-烯炔,并對其進行氟化反應。Wang等[30]已開發出一種溫和有效的可見光誘導的原子轉移自由基加成物1,n-炔烴(n=6,7),與全氟烷基鹵化物發生加成環化,最終生成全氟鹵代N-雜環的方法(圖4)。該方案提供了一種溫和的、完全符合原子經濟學的策略,可以將各種全氟化基團例如CF3I,n-C3F7等對1,6-烯炔,1,7-烯炔進行自由基加成環化。此外,使用帶有叔丁基連接的炔基的1,7-烯炔(n=6,7)進行的反應亦可實現氫原子轉移(HAT)過程的環化,從而產生新的α-鹵代氟代2,4-二氫喹啉化合物。

圖4 可見光催化合成全氟鹵代N-雜環策略[30]Fig.4 The strategy for halogenated perfluoro-N- heterocyclic by blue LEDs[30]
1.2 三氟甲基磺酰氯(CF3SO2Cl)作為三氟甲基試劑
光催化循環的啟動依賴于富電子芳烴的氧化作用,全氟碘代烷烴作為氟化試劑參與反應往往會受到競爭性芳基碘化,從而降低反應效率。因此人們開始尋求新的氟化試劑。2011年,Nagib等[31]發表了將三氟甲基磺酰氯在光照的情況下產生三氟甲基的自由基,并可對未活化的芳烴和雜芳烴進行直接三氟甲基化的工作。在全氟碘代烷烴內部采用缺電子體系(SO2)可以更輕松地還原,與其他氟化試劑相比,三氟甲基磺酰氯相對成本比較低,反應后處理簡單(圖5)。

圖5 可見光催化的雜芳烴的三氟甲基化[31]Fig.5 Trifluoromethylation of heteroarenes by means of photoredox catalysis[31]
隨后利用光催化全氟鹵代烷烴以引發氟烷基自由基的策略被應用到炔烴的含氟雙官能團化的研究中。2017年,Han等[32]開發了一種新的在溫和條件下,利用光氧化還原催化芳基炔烴氯三氟甲基化的反應。該反應利用可以直接購買的CF3SO2Cl作為CF3和Cl源,以高區域選擇性和立體選擇性進行反應。在此機制的反應過程中,CF3自由基和氯離子的產生是通過fac-Ir(ppy)3光催化的CF3SO2Cl還原分解而產生的,通過Suzuki偶聯,可以很容易地將三氟甲磺酰氯轉化為四取代烯烴(圖6)。

圖6 四取代烯烴合成策略及可能的反應機制[32]Fig.6 The strategy of the synthesis of 1,1-bis-arylated olefin and its possible mechanism[32]
香豆素及其衍生物廣泛存在于天然產物以及藥物活性分子骨架中,因其重要的生物學和藥理學特性得到了人們極大的重視[33]。近些年來,科研人員們就香豆素的合成策略已經開發出了形式多樣的方法。2014年,Li等[34]利用Togni試劑制備含氟香豆素,但該方法底物適用性不廣,缺電子底物的產率往往比較低[35]。2018年,Yuan等[36]報道了一種高效的方法,可將CF3SO2Cl作三氟甲基自由基源,與3-芳基丙酸酯反應構建3-三氟甲基香豆素。該反應采用級聯環化/脫芳香化/酯遷移/氧化/再芳香化工藝,在藍光LED燈(16 W)照射下,合成了產物1,為3-三氟甲基香豆素(圖7),產率可高達85%。

圖7 可見光催化合成3-三氟甲基香豆素策略[36]Fig.7 The strategy for the synthesis of 3-trifluoro- methylcoumarin by visible light[36]
筆者課題組一直致力于以綠色化學為主要研究方向。基于前人的可見光催化炔烴的含氟雙官能團化的研究工作基礎,課題組開發出在溫和的條件下,有效地利用光氧化還原催化1,7-烯炔的鹵代三氟甲基化反應方法[36]。該光催化方案提供了一種有效的功能性策略,分別可通過兩種不同的自由基途徑,從產物2,即1,7-烯炔中生成CF3-和含鹵素的喹啉類化合物(圖8)。其中一種自由基途徑是利用有機催化劑9-均三甲苯基-10-甲基吖啶四氟硼酸鹽作為光敏劑,以三氟甲基亞磺酸鈉提供三氟甲基自由基,N-鹵代酞酰亞胺C提供鹵原子,合成喹啉類化合物。另外一種自由基途徑是以fac-Ir(ppy)3作為光敏劑,以三氟甲基磺酰氯作為自由基供體,同時提供三氟甲基自由基和氯自由基。基于機制實驗以及文獻[32],推測該光催化鹵代三氟甲基化反應的初步合理機制可能為:最初,可見光將Mes-Acr+誘導為激發態Mes-Acr+*,從而還原了Langlois試劑。通過單電子轉移(SET)生成CF3自由基A。隨后,CF3自由基被1,7-烯炔的碳碳雙鍵捕獲,得到季碳自由基B,然后進行六元環環化反應,以形成自由基中間體D。隨后,自由基D可以與來自NCP的鹵原子偶合,得到鹵代三氟甲基化產物3。方法2的反應機制與方法1相似,該方法使用具有相同的自由基中間體B和D進行IrIII/IrIV催化。生成的碳正離子被Cl-捕獲,形成目標產物3。

圖8 可見光催化合成喹啉類化合物策略[36]Fig.8 The strategy for the synthesis of quinoline compound by visible light[36]
2 親核性三氟甲基試劑
2.1 Langlois試劑
三氟甲基亞磺酸鈉(Langlois試劑)為固體,反應過程中操作簡單且具有良好的反應性能。2011年,Ji等[37]報道了利用三氟甲基磺酸鈉作為三氟甲基試劑,參與了三氟甲基化反應的工作,該工作為后續的反應研究提供了新的思路。
2017年,Jana等[38]使用光氧化還原催化劑,三氟甲基亞磺酸鈉為氟源,在無金屬條件下,實現1,6-烯炔的加成環化反應(圖9)。該反應通過光誘導三氟甲基磺酸鈉產生三氟甲基自由基,然后進一步與烯烴進行加成、環化,提供乙烯基自由基,該自由基發生電子轉移生成乙烯基陽離子或者被三氟甲基加成的烯烴部分發生電子轉移,形成碳正離子,進行陽離子環化生成乙烯基碳正離子,隨后H2O加成到乙烯基陽離子中,消除H2后生成三氟甲基化的C3-芳酰基/酰化雜環。該工作中一共拓展了43個底物,最高產率可達76%。Jana等[38]還將該方法運用到合成含有三氟甲基基團的藥物分子中,成功得到兩個目標產物。
圖9 藍光催化1,6-烯炔的三氟甲基化和氧化[38]Fig.9 The strategy for the trifluoromethylation and oxidation of 1,6-enynes by blue LEDs[38]
2.2 溴二氟乙酸乙酯試劑
溴二氟乙酸乙酯試劑(CF2CO2Et)由于其獨特的結構而受到人們的重視,因為該部分可以進一步修飾成各種含CF2的官能團。近年來,對過渡金屬介導或過渡金屬催化的芳基鹵化物和芳基硼酸的二氟乙酰化,以構建Csp2-CF2CO2Et鍵的方法得到了深入研究。Ma等[39]等和Caillot等[40]還報道了一種將酰胺,呋喃直接二氟乙酰化的方法。另一種常用策略是利用CF2CO2Et自由基的高反應性。利用可見光催化將該結構加成到雜芳烴[41],烯烴[42],或者異氰酸酯[43]等結構上,這些方法已經被多個研究小組發現并報道。但是,這種光催化策略在炔烴的區域特異性二氟乙酰化中的應用仍然非常有限。
2015年,Fu等[44]開發了一種溫和有效的方法,該方法通過可見光促進炔基與溴代二氟代乙酸乙酯的芳基二氟代乙酰化反應來合成3-二氟代乙酰化香豆素(圖10)。該反應通過串聯自由基環化過程直接形成Csp2-CF2CO2Et和碳碳鍵,實現了通過使用fac-Ir(ppy)3作為催化劑,碳酸鉀作為堿在藍色LED的照射下,2-溴-2,2-二氟乙酸酯和炔酸苯酯反應合成3-二氟乙酰化香豆素的方法。Fu等[44]認為催化循環可能為:首先,將光催化劑[fac-Ir(III)(ppy)3]輻照到激發態[fac-Ir(III)(ppy)3*],將其用BrCF2CO2Et氧化淬滅,生成[fac- Ir(IV)(ppy)3]+配合物和一個·CF2CO2Et自由基物種K。自由基K加成到炔酸酯4中生成自由基中間體H,該中間體進行分子內取代,得到自由基中間體I。中間體I然后被[fac-Ir(IV)(ppy)3]+氧化形成環己二烯基陽離子J并再生[fac-Ir(III)(ppy)3]。最終,通過堿輔助的去質子化得到產物5。

圖10 藍光催化的3-二氟乙酰化香豆素的合成[44]Fig.10 Blue-LEDs-mediated synthesis of 3-difluoroacethlated coumarins[44]
2017年,Li等[45]利用了類似的催化機制,在光催化下,利用9-均三甲苯基-10-甲基吖啶高氯酸鹽代替昂貴的銥催化劑,實現了炔烴的光誘導鄰二氟烷基化和氨基磺酰化。9-均三甲苯基-10-甲基吖啶高氯酸鹽在可見光下催化2-溴-2,2-二氟乙酸乙酯,炔烴和DABCO·(SO2)2與肼的結合,通過自由基加成,并插入SO2進行4個組分反應,得到(E)-乙基 2,2-二氟-4芳基-4-氨磺酰基丁-3-烯酸酯,產率最高可達98%。2018年,該課題組又做出了一部分很有意思的工作,工作內容為采用DABSO作為催化劑在可見光催化下催化合成3-芳基-2-二氟乙酰茚(圖11)[45]。

圖11 光誘導溴二氟乙酸乙酯與1,3-二芳基丙-2-炔-1-酮的反應 [45]Fig.11 A photoinduced reaction of perfluoroalkyl halides with 1,3-diarylprop-2-yn-1-ones[45]
2018年,Deng等[46]同樣利用fac-Ir(ppy)3作為光敏劑,在光催化作用下合成出3-二氟乙酰化喹啉(圖12)。

圖12 N-炔丙基芳胺的光氧化還原催化級聯加成/環化[46]Fig.12 Photoredox-catalyzed cascade addition/cyclization of N-propargyl aromatic amines[46]
3 親電性三氟甲基試劑
3.1 Umemoto試劑
2015年,Tomita等[47]發表了從簡單的炔烴立體選擇性合成四取代的三氟甲基化的烯烴的工作(圖13)。在可見光照射下,[Ir(ppy)2(dtbbpy)](PF6)作為光氧化還原催化劑,用親電的三氟甲基試劑,即Yagupol?skii-Umemoto試劑的三氟甲磺酸鹽,對內部不對稱炔烴進行磺酰氧基三氟甲基化,得到三氟甲基烯基三氟甲磺酸酯,該反應可以很好地控制立體選擇性(以E/Z標記為99∶ 1)。根據一系列的機制實驗,推測出光催化磺酰氧基三氟甲基化的可能反應機制。7與8區域特異性生成α-苯乙烯基型三氟甲基烯基基團6*,該基團通過離域至苯環的p軌道而穩定,關鍵中間體6*可以被高度可氧化的Ir光催化劑(IrIV)氧化為+1e?,氧化為烯烴,從而發生機制類似于先前報道的光氧化還原催化烯烴三氟甲基化的反應,以產生三氟甲基烯基陽離子6+。三氟甲基烯基三氟甲磺酸酯9的高度立體選擇性形成可以用CF3基團的強負電子性來解釋:由于靜電排斥,三氟甲磺酸根陰離子的親核攻擊有利于CF3基團的反式加成。推測可能有另外一種機制,即通過將三氟甲磺酸根陰離子直接添加到6*上,然后進行1e?氧化。

圖13 光氧化還原催化的炔烴立體選擇性轉化為四取代的三氟甲基化烯烴[47]Fig.13 Photoredox-catalyzed stereoselective conversion of alkynes into tetrasubstituted trifluoromethylated alkenes[47]
2018年,Malpani等[48]發表了采用Umemoto試劑提供CF3和H2O直接氧化成炔烴的工作,主要內容是利用光氧化還原催化反應,通過快速的烯醇-酮互變異構反應,獲得α-三氟甲基酮(圖14)。該反應表現出高的官能團耐受性和區域選擇性。該方案獲得的α-CF3取代的二酮合成了各種含CF3的雜環結構,從而證明了該方法廣泛的適用范圍。該策略同樣被應用在一種光氧化還原催化的分子內環化反應,以用于合成三氟甲基化的雜環化合物[49]。該反應在溫和的光催化條件下,官能團耐受性強,可以制備不同的含氧、硫或氮的雜環,底物廣泛的適用范圍證明了該策略在提高底物復雜性方面的潛力。

圖14 光氧化還原催化的三氟甲基化分子內環化[49]Fig.14 Photoredox-catalyzed trifluoromethylative intramolecular cyclization[49]
3.2 Togni試劑
Xiang等[50]也應用過類似的策略,在可見光照射下,2-乙炔基苯磺酰胺在光催化劑存在下與Togni試劑反應,以良好的收率生成3-(2,2,2-三氟亞乙基)-2,3-二氫苯并[d]異噻唑1,1-二氧化物,隨后的C—N鍵形成會產生相應的產物(圖15)。

圖15 在可見光下通過2-乙基苯磺酰胺的三氟甲基化 反應生成苯并磺酰胺[50]Fig.15 Generation of benzosultams via trifluoro- methylation of 2-ethynylbenzensulfonamide under visible light[50]
4 結語
綜上所述,從對可見光催化炔烴的含氟雙官能團化的研究進展總結中可以看出,目前常用的光催化劑主要為貴金屬配合物(Ir、Ru等)和有機染料,金屬配合物或者含有大π離域結構的有機分子在吸收低能量的可見光后,激發發生分子內電荷轉移(CT),從而引發光反應。這些貴金屬配合物和有機染料光催化劑大多結構復雜、不易合成、成本高昂,難以實現大規模的產業化應用。因此,追求更加廉價實用的光催化反應體系勢必會對擴展其應用帶來變革。該方向的發展總趨勢是催化劑由多吡啶配體釕催化劑、多吡啶配體銥催化劑向廉價的有機染料或者鋅、銅等價格廉價易得的金屬催化劑轉化,同時氟源的來源逐漸簡化,合成的目標產物更傾向于直接合成藥物分子并逐步趨向于工業化。
