甲苯磺酸艾多沙班異構體雜質的合成
2021-04-07 12:23:18王子月
浙江化工 2021年3期
徐 進,王子月
(江蘇華陽制藥有限公司,江蘇 宿遷 223700)
2015 年1 月8 日,由日本第一三共公司開發的艾多沙班對甲苯磺酸鹽一水合物(edoxaban tosylate monohy—drate,商品名:Savaysa)經美國食品藥品監督管理局(FDA)批準上市。甲苯磺酸艾多沙班(EDXB)是日本上市的首個口服給藥的抗凝藥物,在全球也是首次問世。EDXB 為直接FXa抑制劑,而FXa 正是當今開發新一代抗凝藥物的主要靶點。口服給藥為臨床用藥提供方便,臨床試驗驗證了本品安全有效。EDXB 將在抗凝藥物領域占據更多的優勢,從而獲得更多的商業機會。
甲苯磺酸艾多沙班,化學名為N1-(5-氯吡啶-2-基)-N2-((1S,2R,4S)-4-((二甲氨基)羰基)-2-(((5-甲基-4,5,6,7-四氫噻唑并[5,4-c]吡啶-2-基)甲酰基)氨基)環己基)乙二酰胺對甲苯磺酸一水合物,EDXB(1S,2R,4S)的結構式見圖1。
EDXB 藥物的合成以及存放過程易產生雜質,對藥品質量產生嚴重的影響。EDXB 對映異構體和差向異構體是主要的合成工藝雜質,EDXB對映異構體即甲苯磺酸艾多沙班(1R,2S,4R)的結構式見圖2(a)。EDXB 非對映(差向)異構體的結構式有兩種,即甲苯磺酸艾多沙班(1R,2R,4S)和甲苯磺酸艾多沙班(1S,2S,4R),結構式見圖2(b)和圖2(c)。
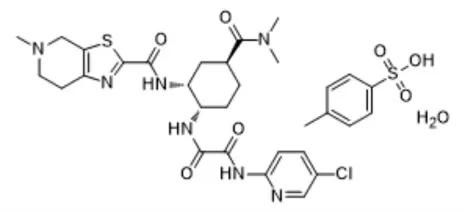
圖1 甲苯磺酸艾多沙班(1S,2R,4S)的結構式
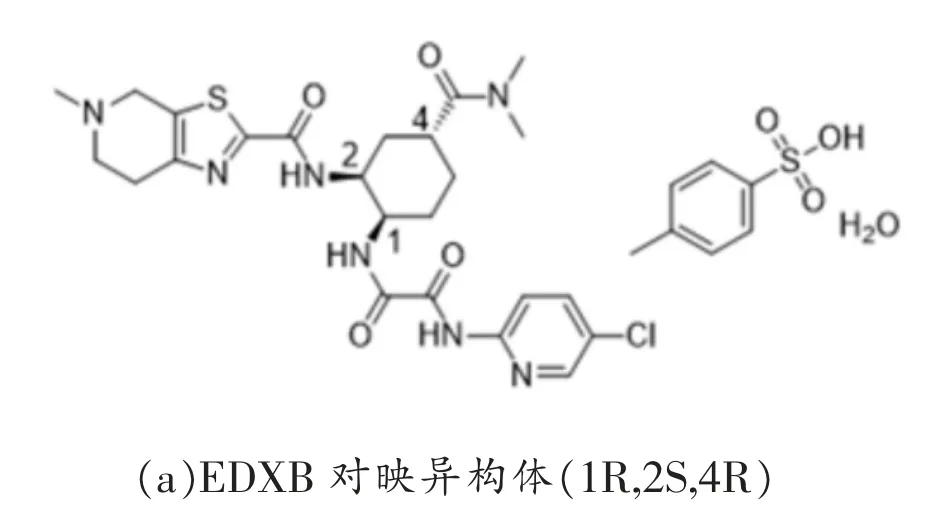
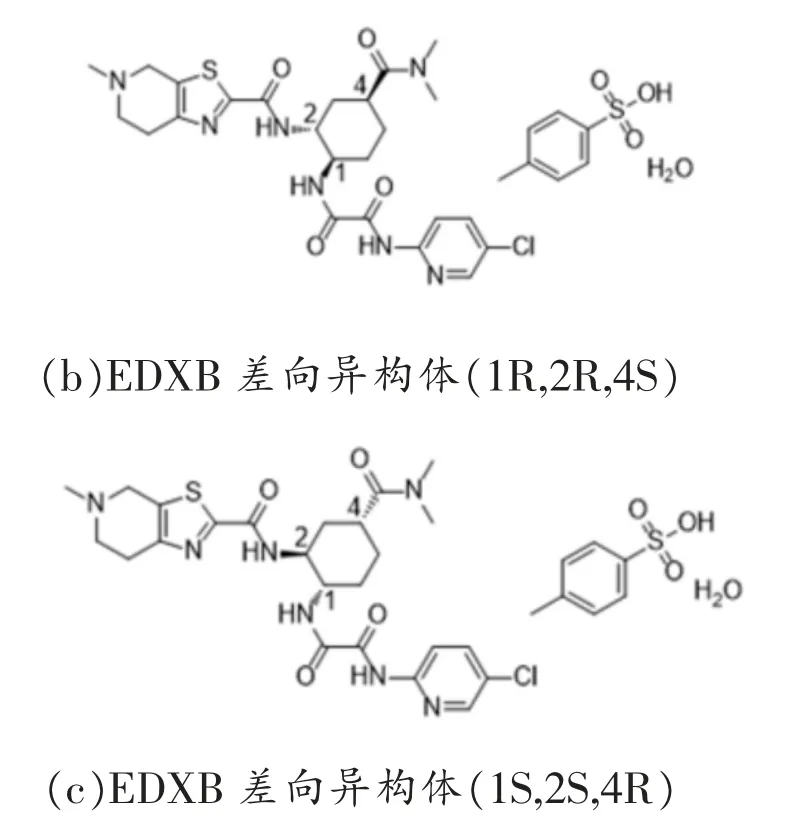
圖2 EDXB 的異構體
Nagata 等[1]報道了以3-環己烯-1-甲酸為起始物料合成疊氮化合物的合成路線,見圖3。Toshiharu 等[2]、太田敏晴等[3]和Yoshikawa 等[4]均報道了疊氮化合物通過水解還原后與主要的結構片段縮合生成EDXB 的工藝。
現有工藝得到的都是單一構型的EDXB,本文通過不同構型的苯基乙胺與3-環己烯-1-甲酸成鹽并水解,得到不同構型的3-環己烯-1-甲酸,進而得到不同構型的環氧化合物。環氧化合物通過氨解、柱層析后可以得到4 個不同構型的氨解產物,重氮化后可以得到4 個不同構型的疊氮化合物,再經過后續的反應步驟得到EDXB 和三個異構體雜質,具體合成路線見圖4。
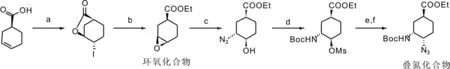
圖3 以3-環己烯-1-甲酸為起始物料合成疊氮化合物
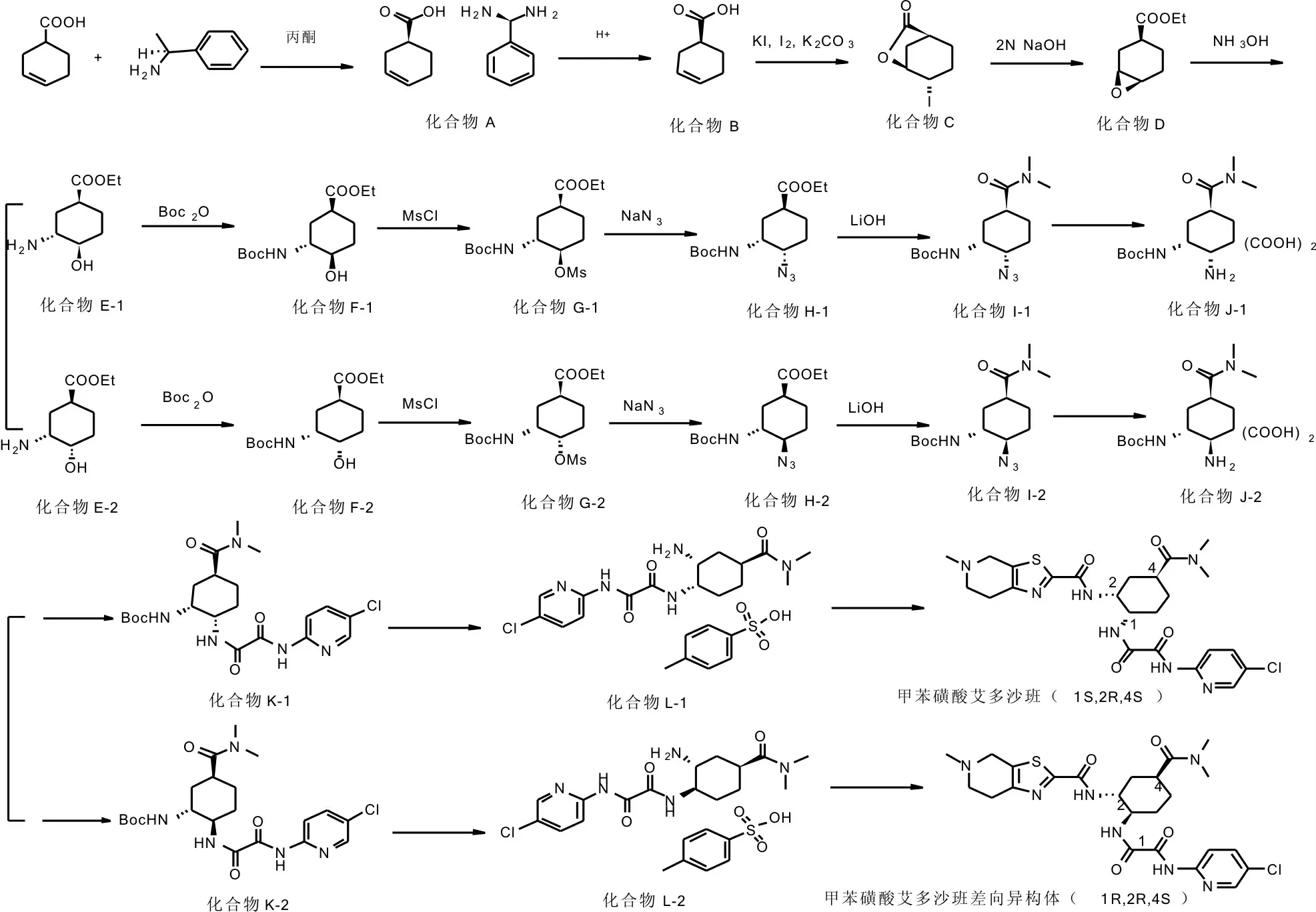

圖4 EDXB 和三個異構體雜質的合成路線
1 實驗部分
1.1 甲苯磺酸艾多沙班差向異構體(1R,2R,4S)的制備
(1)將200 g 3-環己烯-1-甲酸加至1 L 丙酮中,回流溶解1 h,將192.1 g s-苯基乙胺用0.5 L丙酮溶解滴加到反應液中,緩慢降溫,降至40 ℃反應2 h,然后降溫至20 ℃~25 ℃,抽濾,濾餅在40 ℃~45 ℃下鼓風干燥得到270 g 固體,加入8倍丙酮重結晶,然后降溫至20 ℃~25 ℃析晶,抽濾,濾餅在40 ℃~45 ℃下鼓風干燥得到170 g 化合物A。
(2)將170 g 化合物A 加至3 L 二氯甲烷中,加入3 L 2 mol/L 的HCl 溶液攪拌2 h,分液,水層用2 L 二氯甲烷反萃,有機層用3 L 飽和氯化鈉溶液洗滌,用100 g 無水硫酸鈉干燥,在40 ℃~45℃下濃縮至干,得到108 g 化合物B。
(3)將108 g 化合物B 加至2 L 二氯甲烷中,加入178 g KI、90 g 碳酸氫鈉、2 L 純化水,降溫攪拌,溫度降至5 ℃~10 ℃,加入283 g 碘,升溫至20 ℃~25 ℃攪拌4 h,溫度降至5 ℃~10 ℃加入1 mol/L 硫代硫酸鈉溶液,加入2 L 二氯甲烷分液,水層用1 L 二氯甲烷反萃,合并有機層,分別用飽和碳酸氫鈉、純化水、飽和氯化鈉洗滌,用100 g 無水硫酸鈉干燥,濃縮至干,得到145 g 化合物C。
(4)將145 g 化合物C 加至2 L 乙醇中,加入350 mL 的4 mol/L 氫氧化鈉水溶液,在25 ℃~30℃下攪拌4 h,在35 ℃濃縮得到油狀物,加入1 L二氯甲烷萃取分液,用100 g 無水硫酸鎂干燥,抽濾,濃縮濾液至干,得到80 g 化合物D。
(5)將80 g 化合物D 加至4 L 乙醇中,加入800 mL 氨水,在40 ℃~45 ℃下攪拌24 h,在40℃濃縮,加入250 mL 乙醇,攪拌,將100 mL Boc(二碳酸二叔丁酯)用乙醇溶解滴加到反應液中,滴加完畢,在20 ℃~25 ℃下攪拌2 h,濾液濃縮至干,得到化合物F-1 和F-2 的混合物,然后過硅膠柱(PE 和EA 梯度洗脫)得到50 g 化合物F-1及15 g 化合物F-2。
(6)將15 g 化合物F-2 加至500 mL 二氯甲烷中,加入三乙胺降溫攪拌,溫度降至0 ℃滴加18 g MsCl,滴加完畢,在20 ℃~25 ℃下攪拌2 h,溫度降至0 ℃,滴加250 mL 0.5 mol/L 鹽酸,滴加完畢,加入500 mL 二氯甲烷萃取分液,有機層用1 L 純化水、1 L 飽和氯化鈉洗滌,50 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體化合物G-2,將G-2 加至1 L DMF 中,加入23 g 疊氮化鈉,在65 ℃~75 ℃下攪拌24 h,溫度降至25℃,加入2 L 二氯甲烷、5 L 純化水萃取分液,水層用2 L 二氯甲烷反萃,有機層用2 L 飽和氯化鈉洗滌,50 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后過硅膠柱,用(PE:EA=20:1)沖洗,得到9 g 化合物H-2。
(7)將9 g 化合物H-2 加至500 mL 二氯甲烷、100 mL 純化水中,加入1.5 g 氫氧化鋰一水合物,在25 ℃~30 ℃下攪拌24 h,用1 mol/L 氯化銨溶液調反應液pH 至6,加入1.5 L 乙酸乙酯分液,用1.5 L 飽和氯化鈉洗滌乙酸乙酯,50 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后加入1 L 二氯甲烷、2.5 g 鹽酸二甲胺、3.6 g EDCI、2.7 g HOBT、N-甲基嗎啉,在35 ℃~45 ℃下攪拌24 h,加入飽和碳酸氫鈉溶液分液,用50 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后過硅膠柱,用(MEOH:DCM=1:50)沖洗,得到4.5 g 化合物I-2。
(8)將化合物I-2 加至500 mL 乙醇、1.5 g 甲酸銨、0.5 g 鈀碳中,攪拌4 h,抽濾,濾液濃縮至干得到油狀物,然后加入60 mL 乙腈溶解,在25 ℃~30 ℃下攪拌,加入1.1 g 草酸攪拌3 h,抽濾,濾餅于40 ℃~45 ℃下鼓風干燥得到3.9 g 化合物J-2。
(9)將3.9 g 化合物J-2、2-[(5-氯吡啶)氨基]-2-氧代乙酸乙酯鹽酸鹽、三乙胺加入DMF 中,反應10 h,干燥得到4.2 g 固體化合物K-2。
(10)將4.2 g 化合物K-2 加入四氫呋喃中,加入對甲苯磺酸一水合物,反應得到4.5 g 化合物L-2。
(11)將4.5 g 化合物L-2 與乙腈、三乙胺、縮合試劑、5-甲基-4,5,6,7-四氫噻唑并[5,4-c]吡啶-2-羧酸鹽酸鹽反應,再與甲苯磺酸一水合物反應,得到2.8 g 目標化合物,即甲苯磺酸艾多沙班差向異構體(1R,2R,4S)。
1.2 甲苯磺酸艾多沙班對映異構體(1R,2S,4R)的制備
(1)將200 g 3-環己烯-1-甲酸加至1 L 丙酮中,回流溶解1 h,將192.1 g R-苯基乙胺用0.5 L丙酮溶解,滴加到反應液中,緩慢降溫,降至40 ℃反應2 h,然后降溫至20 ℃~25 ℃,抽濾,濾餅在40 ℃~45 ℃下鼓風干燥,得到282 g 固體,加入8倍丙酮重結晶,然后降溫至20 ℃~25 ℃析晶,抽濾,濾餅在40 ℃~45 ℃下鼓風干燥得到173 g 化合物A-1。
(2)將173 g 化合物A-1 加至3 L 二氯甲烷中,加入3 L 的2 mol/L 的HCl 溶液攪拌2 h,分液,水層用2 L 二氯甲烷反萃,有機層用3 L 飽和氯化鈉溶液洗滌,用100 g 無水硫酸鈉干燥,在40 ℃~45 ℃下濃縮至干,得到112 g 化合物B-1。
(3)將112 g 化合物B-1 加至2 L 二氯甲烷中,加入180 g KI、90 g 碳酸氫鈉、2 L 純化水,降溫攪拌,溫度降至5 ℃~10 ℃,加入285 g 碘,升溫至20 ℃~25 ℃,攪拌4 h,溫度降至5 ℃~10 ℃加入1 mol/L 硫代硫酸鈉溶液,加入2 L 二氯甲烷分液,水層用1 L 二氯甲烷反萃,合并有機層,分別用飽和碳酸氫鈉、純化水、飽和氯化鈉洗滌,用110 g 無水硫酸鈉干燥,濃縮至干,得到化合物C-1。
(4)將150 g 化合物C-1 加至2.1 L 乙醇中,加入360 mL 4 mol/L 氫氧化鈉水溶液,在25 ℃~30 ℃下攪拌4 h,在35 ℃濃縮得到油狀物,加入1 L 純化水及1 L 二氯甲烷萃取分液,用100 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到82 g 化合物D-1。
(5)將82 g 化合物D-1 加至4 L 乙醇中,加入800 mL 氨水,在40 ℃~45 ℃下攪拌24 h,在40 ℃濃縮,加入250 mL 乙醇,攪拌,將100 mL Boc 用乙醇溶解,滴加到反應液中,滴加完畢在20 ℃~25 ℃下攪拌2 h,濾液濃縮至干,得到化合物F-3 和F-4 的混合物,然后過硅膠柱(PE 和EA 梯度洗脫)得到14 g 化合物F-3 及46 g 化合物F-4。
(6)將46 g 化合物F-4 加至450 mL 二氯甲烷中,加入三乙胺降溫攪拌,溫度降至0 ℃,滴加54 g MsCl,滴加完畢在20 ℃~25 ℃下攪拌2 h,溫度降至0 ℃,滴加800 mL 0.5 mol/L 鹽酸,滴加完畢加入450 mL 二氯甲烷萃取分液,有機層用0.9 L 純化水、0.9 L 飽和氯化鈉洗滌,100 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體化合物G-4,將G-4 加至0.9 L DMF 中,加入疊氮化鈉在65 ℃~75 ℃下攪拌24 h,溫度降至25 ℃,加入1.8 L 二氯甲烷、4.5 L 純化水萃取分液,水層用1.8 L 二氯甲烷反萃,有機層用1.8 L 飽和氯化鈉洗滌,100 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后過硅膠柱,用(PE:EA=20:1)沖洗,得到28 g 化合物H-4。
(7)將28 g 化合物H-4 加至460 mL THF、95 mL 純化水中,加入4 g 氫氧化鋰一水合物,在25℃~30 ℃下攪拌24 h,用1 mol/L 氯化銨溶液調節反應液pH 至6,加入3 L 乙酸乙酯分液,用2 L飽和氯化鈉洗滌乙酸乙酯,100 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后加入0.9 L 二氯甲烷、8 g 鹽酸二甲胺、12 g EDCI、9 g HOBT、N-甲基嗎啉,在35 ℃~45 ℃下攪拌24 h,加入飽和碳酸氫鈉溶液分液,100 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后過硅膠柱,用(MEOH:DCM=1:50)沖洗,得到13 g 化合物I-4。
(8)將13 g 化合物I-4 加至450 mL 乙醇、5 g甲酸銨、2 g 鈀碳中,攪拌4 h,抽濾,濾液濃縮至干,得到油狀物,然后加入60 mL 乙腈溶解,在25℃~30 ℃下攪拌,加入3.5 g 草酸攪拌3 h,抽濾,濾餅在40 ℃~45 ℃下鼓風干燥,得到11.5 g 固體化合物J-4。
(9)將11.5 g 化合物J-4、2-[(5-氯吡啶)氨基]-2-氧代乙酸乙酯鹽酸鹽、三乙胺加入DMF中,反應10 h,干燥得到13.1 g 固體化合物K-4。
(10)將13.1 g 化合物K-4 加入四氫呋喃中,加入對甲苯磺酸一水合物,反應得到14.5 g 固體化合物L-4。
A4(尾聲,見例9)在寫法上仍沿用了第一主題的旋律,但是情緒上有較大變化。鋼琴Ⅰ需要控制好尾聲在力度、速度、音色上的變化,與鋼琴Ⅱ形成呼應,并隨著鋼琴Ⅱ在節奏時值上的放慢,力度逐漸減弱直至聲音的消失。第126至127小節的演奏要從容富有幻想。
(11)將14.5 g 固體化合物L-4 與乙腈、三乙胺、縮合試劑、5-甲基-4,5,6,7-四氫噻唑并[5,4-c]吡啶-2-羧酸鹽酸鹽反應,再與甲苯磺酸一水合物反應得到7.5 g 目標化合物,即甲苯磺酸艾多沙班對映異構體(1R,2S,4R)。
1.3 甲苯磺酸艾多沙班差向異構體(1S,2S,4R)的制備
步驟(1)~(5)與甲苯磺酸艾多沙班對映異構體(1R,2S,4R)制備方法相同。
(6)將14 g 化合物F-3 加至500 mL 二氯甲烷中,加入三乙胺降溫攪拌,溫度降至0 ℃滴加56 g MsCl,滴加完畢,在20 ℃~25 ℃下攪拌2 h,溫度降至0 ℃,滴加800 mL 0.5 mol/L 鹽酸,滴加完畢,加入500 mL 二氯甲烷萃取分液,有機層用1 L 純化水、1 L 飽和氯化鈉洗滌,50 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體化合物G-3,將G-3 加至1 L DMF 中,加入23 g 疊氮化鈉,在65 ℃~75 ℃下攪拌24 h,溫度降至25℃,加入2 L 二氯甲烷、5 L 純化水萃取分液,水層用2 L 二氯甲烷反萃,有機層用2 L 飽和氯化鈉洗滌,100 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后過硅膠柱,用(PE:EA=20:1)沖洗,得到8.7 g 化合物H-3。
(7)將8.7 g 化合物H-3 加至480 mL THF、100 mL 純化水中,加入氫氧化鋰一水合物,在25℃~30 ℃下攪拌24 h,用1 mol/L 氯化銨溶液調節反應液pH 至6,加入3 L 乙酸乙酯分液,用2 L飽和氯化鈉洗滌乙酸乙酯,50 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后加入1 L 二氯甲烷、2.5 g 鹽酸二甲胺、3.6 g EDCI、2.7 g HOBT、N-甲基嗎啉,在35 ℃~45 ℃下攪拌24 h,加入飽和碳酸氫鈉溶液分液,用50 g 無水硫酸鎂干燥,抽濾,濾液濃縮至干,得到淡黃色固體,然后過硅膠柱,用(MEOH:DCM=1:50)沖洗,得到4.1 g 化合物I-3。
(8)將4.1 g 化合物I-3 加至500 mL 乙醇、1.5 g 甲酸銨、1.5 g 鈀碳中,攪拌4 h,抽濾,濾液濃縮至干,得到油狀物,然后加入60 mL 乙腈溶解,在25 ℃~30 ℃下攪拌,加入1.1 g 草酸攪拌3 h,抽濾,濾餅在40 ℃~45 ℃下鼓風干燥,得到3.6 g 固體化合物J-3。
(9)將3.6 g 固體化合物J-3、2-[(5-氯吡啶)氨基]-2-氧代乙酸乙酯鹽酸鹽、三乙胺加入DMF中,反應10 h,干燥得到3.8 g 化合物K-3。
(10)將3.8 g 化合物K-3 加入四氫呋喃中,加入對甲苯磺酸一水合物,反應得到4.1 g 固體化合物L-3。
2 結果與雜質控制
2.1 甲苯磺酸艾多沙班差向異構體雜質液相檢測及結構確證
2.1.1 液相分離及純度檢測
采用高效液相色譜法(中國藥典2020 年版四部通則0512)測定EDXB 差向異構體雜質,色譜條件:十八烷基硅烷鍵合硅膠為填充劑(YMCPack ODS-AM,150 mm×4.6 mm,3 μm 或效能相當的色譜柱);以2.5 g/L 辛烷磺酸鈉水溶液(用磷酸調pH 至2.2)為流動相A,乙腈為流動相B 進行液相測定;進樣體積為10 μL。EDXB 差向異構體雜質與EDXB 分離圖譜見圖5。
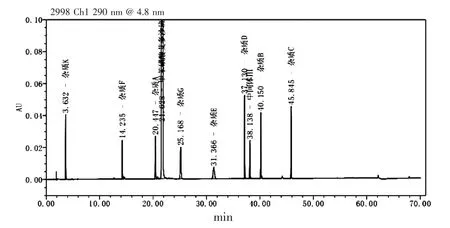
圖5 EDXB 差向異構體雜質與EDXB 液相分離圖
EDXB 差向異構體(1R,2R,4S)和差向異構體(1S,2S,4R)在色譜條件下同一位置出峰(保留時間為25.16 min),即雜質G,EDXB 保留時間為21.62 min。兩個差向異構體雜質合并控制,限度為0.15%,EDXB 差向異構體(1R,2R,4S)和差向異構體(1S,2S,4R)液相色譜純度分別為98.1%和98.2%。
2.1.2 甲苯磺酸艾多沙班差向異構體雜質結構確證
甲苯磺酸艾多沙班差向異構體(1R,2R,4S)核磁共振檢測數據:1H NMR (400 MHz,DMSO-d6,δ):1.458~1.549 (1H,d),1.668~1.791 (3H,m),2.034~2.130 (2H,m),2.270 (3H,s),2.789 (3H,s),3.002(7H,m),3.372(2H,s),3.656(2H,s),3.986~4.056 (1H,m),4.445~4.617 (3H,d),7.089~7.109(2H,d),7.451~7.471(2H,d),7.987~8.044(2H,m),8.444~8.452 (1H,m),8.745~8.763 (1H,d),9.197~9.215(1H,d),10.272(2H,s)。13C NMR(400 MHz,DMSO~d6,δ):21.17,24.09,25.30,27.15,31.81,33.41,35.50,37.00,42.29,48.45,50.02,50.82,51.01,115.48,125.87,126.22,127.20,128.71,138.79,138.83,144.96,147.30,148.26,148.86,158.66,159.22,160.08,163.69,174.55。
甲苯磺酸艾多沙班差向異構體(1S,2S,4R)核磁共振檢測數據:1H NMR (400 MHz,DMSO~d6,δ):1.458~1.549 (1H,d),1.670~1.793 (3H,m),2.038~2.135 (2H,m),2.266 (3H,s),2.789 (3H,s),3.004(7H,m),3.380(2H,s),3.659(2H,s),3.9868~4.059(1H,m),4.449~4.625(3H,d),7.087~7.107(2H,d),7.451~7.471 (2H,d),7.984~8.045 (2H,m),8.440~8.446 (1H,m),8.746~8.765 (1H,d),9.205~9.223(1H,d),10.267(2H,s)。13C NMR(400 MHz,DMSO~d6,δ):21.19,24.11,25.35,27.17,31.83,33.41,35.47,36.99,42.27,48.53,50.02,50.82,50.97,115.45,125.88,126.22,127.23,128.66,138.64,138.79,145.23,147.32,148.29,148.91,158.70,159.20,160.07,163.73,174.41。
2.2 甲苯磺酸艾多沙班對映異構體雜質液相檢測及結構確證
2.2.1 液相分離及純度檢測
采用高效液相色譜法(中國藥典2020 年版四部通則0512)測定EDXB 對映異構體。色譜條件:以IA 為色譜柱(CHIRALPAK IA 4.6 mm×250 mm,5 μm 或效能相當的色譜柱);以無水乙醇-乙腈(50:50)為流動相,進樣體積為10 μL。EDXB 對映異構體雜質與EDXB 分離圖譜見圖6。
色譜圖中對映異構體峰(保留時間為16.99 min)即雜質H,EDXB 保留時間為23.52 min,按自身對照法計算含量,對映異構體不得超過0.15%。EDXB 對映異構體液相色譜純度為97.3%。
圖6 EDXB 對映異構體雜質與EDXB 液相分離圖譜
2.2.2 甲苯磺酸艾多沙班對映異構體雜質結構確證
甲苯磺酸艾多沙班對映異構體(1R,2S,4R)核磁共振檢測數據:1H NMR(400 MHz,DMSO~d6,δ):1.458~1.549 (1H,d),1.668~1.791 (3H,m),2.034~2.130(2H,m),2.270(3H,s),2.789(3H,s),3.002(7H,m),3.372(2H,s),3.654(2H,s),3.986~4.056(1H,m),4.445~4.626 (3H,d),7.089~7.109 (2H,d),7.451~7.471(2H,d),7.987~8.044(2H,m),8.444~8.452(1H,m),8.744~8.763(1H,d),9.196~9.215(1H,d),10.272(2H,s)。13C NMR (400 MHz,DMSO~d6,δ):21.18,24.10,25.33,27.15,31.81,33.41,35.48,36.99,42.27,48.50,50.02,50.82,50.98,115.47,125.88,126.22,127.21,128.67,138.69,138.79,145.17,147.32,148.28,148.90,158.69,159.21,160.07,163.72,174.36。
3 結論
本文制備了高純度的EDXB 對映異構體和差向異構體作為對照品,產物結構均經1H NMR和13C NMR 確證,且通過合適的液相色譜條件的篩選,EDXB 對映異構體、差向異構體和EDXB 本身得到了很好的分離,從而控制EDXB 的藥品質量。本方法制備的EDXB 差向異構體雜質(1R,2R,4S) 的純度可以達到98.1%,EDXB 差向異構體雜質(1S,2S,4R)的純度可達到98.2%,EDXB 對映異構體雜質(1R,2S,4R)的純度可以達到97.3%,為甲苯磺酸艾多沙班原料藥及相關制劑的工藝研究和質量控制提供了有效的保障。
