水稻多胚基因CRISPR/Cas9基因編輯載體的構建
2021-03-15 06:50:36陳強何勇田志宏
南方農業·上旬 2021年2期
陳強 何勇 田志宏
摘 要 為構建水稻多胚基因OsPE的CRISPR/Cas9基因編輯載體,以OsU6a啟動子為模板,通過兩輪Overlapping PCR構建出一個sgRNA的表達盒。在T4連接酶的作用下,將sgRNA表達盒連接到酶切后的CRISPR/Cas9編輯載體中。先進行菌液PCR驗證和酶切驗證,后通過測序檢測,獲得一套多胚基因的pYLCRISPR/Cas9-gRNA載體,為多胚基因的進一步研究提供新的路徑。
關鍵詞 水稻多胚基因;CRISPR/Cas9;基因編輯;載體構建
正常情況下水稻有性生殖的過程中只產生一個胚囊,一個胚囊只含有一個受精卵,受精后也只能發育一個胚[1]。通過自然進化產生的一些多胚性水稻,其種子無規律地分布在穗子中,且其不能夠穩定遺傳。水稻多胚基因OsPE是一個功能獲得性的基因,位于水稻的第3條染色體上,cDNA全長2 755 bp,含有2個外顯子,到目前為止,在水稻基因組中未發現OsPE的同源基因[2]。多胚基因在水稻雜交和加速育種等方面都有著重要的應用,種子由多到少,生殖由單一胚到二個胚乃至多個胚,可用于水稻多倍體的研究,通過加倍獲得純合的多倍體種子,加強育種優勢,縮短育種需要的時間,同時可以節省母種、提高產量、改良品種等。發掘多胚基因的遺傳機理,改良多胚基因的遺傳穩定性是多胚基因發揮應用價值的重要前提。
基因組編輯技術是近幾年興起的一項能對基因組進行精確修飾的技術,通過使用核酸內切酶在基因組中可完成堿基的替換、插入、彌補小片段缺失等[3-4]。此外,具有轉錄抑制因子或激活因子的CRISPR/Cas9系統的修改版本,允許對靶基因進行強有力的轉錄抑制或激活[5]。CRISPR/Cas9主要由single-guide RNA和其引導的Cas9蛋白兩部分組成,CRISPR/Cas9技術不僅適用性廣,還可以同時進行多重靶向的編輯,且脫靶率比其他編輯方式低。近年來,基因編輯技術在水稻育種方面得到了大量應用,借助基因編輯的手段,通過T-DNA的插入,使基因序列發生一定的突變,能夠獲得一些穩定遺傳的植物材料。
1 材料與方法
1.1 實驗材料
供試大腸桿菌菌株DH10B和根癌農桿菌EHA105由本實驗室保存,pYLCRISPR/Cas9基因編輯系統由華南農業大學劉耀光教授惠贈。實驗中使用的高保真酶、質粒提取試劑盒、凝膠回收試劑盒等購于南京諾唯贊生物科技有限公司。
1.2 靶點選擇及引物設計
根據在NCBI中公布的水稻基因組序列,查找多胚基因OsPE的序列。利用在線軟件CRISPR-P分析多胚基因的靶點特異性,在ORF功能結構區域篩選挑選合適的靶點,靶點的GC含量不低于40%,選擇合適的Cas9靶點序列為T1:TGCTCTGTCTGAGTAAAGCA。利用Primer-5軟件設計編輯引物,引物由擎科(武漢)生物科技有限公司合成,純化級別PAGE。依據靶位點序列及sgRNA構建方法選擇引物(見表1)。
1.3 sgRNA表達盒的構建
以pYLsgRNA-OsU6a/LacZ載體質粒為模板,通過兩步Overlapping PCR往sgRNA表達盒中導入靶序列。第1步使用高保真DNA聚合酶構建一個10 μL的反應體系,在這個反應中使用4種引物,U-F和gR-R各0.5 μL,gRT1和OsU6aT1各0.25 μL。反應進行25個循環,95 ℃變性15 s;60 ℃退火15 s;72 ℃延伸45 s。第2步使用2種引物,Pps-GGL和Pgs-GGR各0.4 μmol·L-1,反應程序同上。對反應產物進行凝膠檢測和測序檢測。本實驗表達盒由來源于水稻的RNA啟動子U6a啟動。
1.4 重組載體的構建與轉化
利用限制性核酸內切酶BsaI識別位點與切割位點不重疊的特性,采用Golden gate cloning法構建雙元載體[6-7]。選擇適用于單子葉植物的pYLCRISPR/Cas9-Pubi-H載體,pYLCRISPR/Cas9質粒使用BsaI酶進行酶切,1 μg的質粒反應30 min,反應溫度37 ℃,獲得線性化載體。然后T4 DNA ligase對pYLCRISPR/Cas9質粒和上述gRNA表達盒進行連接,利用Golden gate cloning法將其克隆至pYLCRISPR/Cas9的BsaI位點。反應體系見表2。
反應程序:10 ℃反應5 min,20 ℃反應5 min,共進行15個循環。將重組產物用熱激法轉化至感受態大腸桿菌DH10B中,37 ℃平板涂布放置過夜,重組的載體質粒中含有卡那抗性基因,用含有50 mg·L-1的LB培養基篩選陽性菌落,也可以加入X-gal和IPTG進行藍白斑篩選。
1.5 轉化子的檢測
挑取平板上的單克隆菌擴大培養,同時對菌落進行PCR檢測,菌落PCR中使用的引物為CRISPRCX-F和CRISPRCX-R。反應程序:94 ℃預處理90 s,94 ℃高溫變性20 s,56 ℃退火20 s,72 ℃片段延伸90 s,最后延伸5 min,共進行30個循環。凝膠電泳檢測結果為陽性克隆,對陽性克隆分別進行2個靶點特異性引物CRISPRCX-F/OsU6aT1和gRT1/CRISPRCX-R的檢測,PCR的反應程序與上面相同,檢測陽性克隆的插入方向。挑選部分陽性克隆重組菌提取質粒,將重組質粒送擎科(武漢)生物科技有限公司進行測序,測序引物同上檢測引物。
2 結果與分析
2.1 sgRNA表達盒的獲取
用Overlapping PCR法通過特異性引物對pYLsgRNA-OsU6a/LacZ啟動子質粒進行擴增,經過兩輪PCR擴增,對第二輪PCR產物進行凝膠電泳,獲得片段大小應為831 bp,凝膠電泳結果與預期相符(見圖1),部分顯示無條帶可能與第二輪PCR底物濃度有關。
2.2 表達載體的獲得與驗證
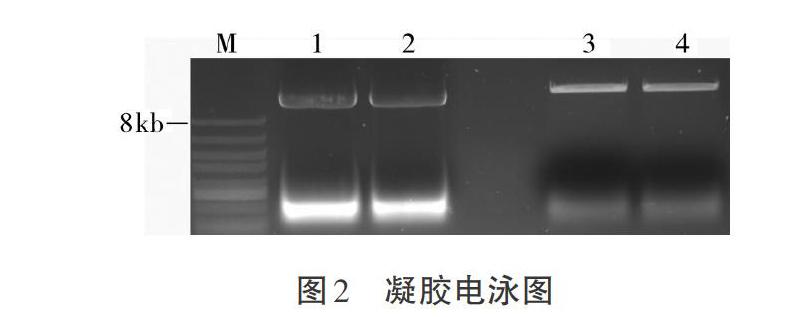
對PCR產物直接進行回收,利用Golden gate cloning法將其連接到BsaI酶切后的載體CRISPR/Cas9中。提取轉化菌的質粒,用MiuI酶切重組質粒鑒定是否連接成功,結果顯示切出與預期大小相同的目的片段,表明已成功連接(見圖2)。
CRISPR/Cas9系統有兩個主要組成部分,其中一個是單一的引導RNA (sgRNA),它識別特定的DNA序列,以及在目標位點產生DSB的Cas9蛋白[8-10],因此在改變sgRNA的設計時,可以靈活地選擇多種位點。本研究中跟Cas9相關的基因由Pubi-H啟動子驅動,而sgRNA則是由來源于水稻的U6a驅動[11],以確保轉錄出的特異性單導RNA能夠進入細胞核中與Cas9蛋白結合,構建的載體中用于水稻材料篩選的抗生素是潮霉素。載體圖譜見圖3,根據CRISPR/Cas9載體圖譜改良。
2.3 轉化菌的PCR檢測
重組質粒轉化DH10B后,挑取呈現出藍斑的單菌落置于搖床放大培養,對陽性的單菌落進行PCR檢測,使用引物CRISPRCX-F和CRISPRCX-R,得到大小亮度適合的片段,符合預期(見圖4)。
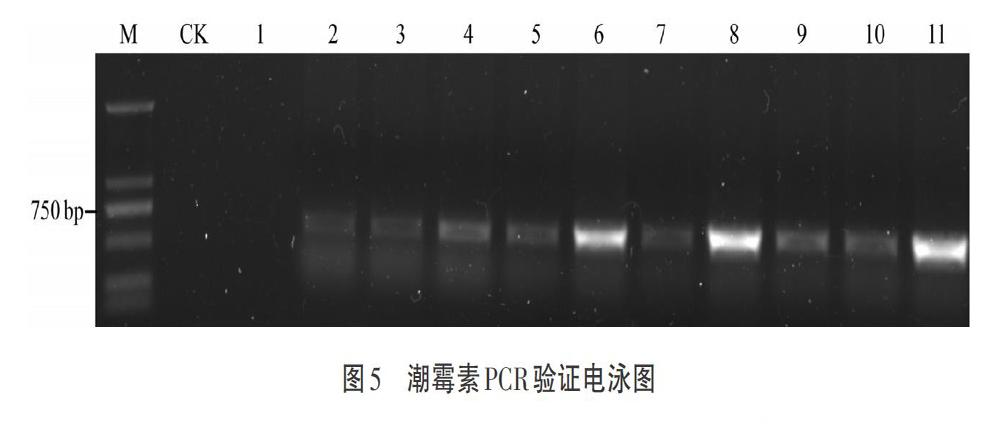
同時對陽性單克隆進行一個潮霉素的PCR驗證,得到大小約750 bp的片段,2號孔至11號孔都克隆出條帶,表明部分表達載體成功轉化進入大腸桿菌(見圖5)。
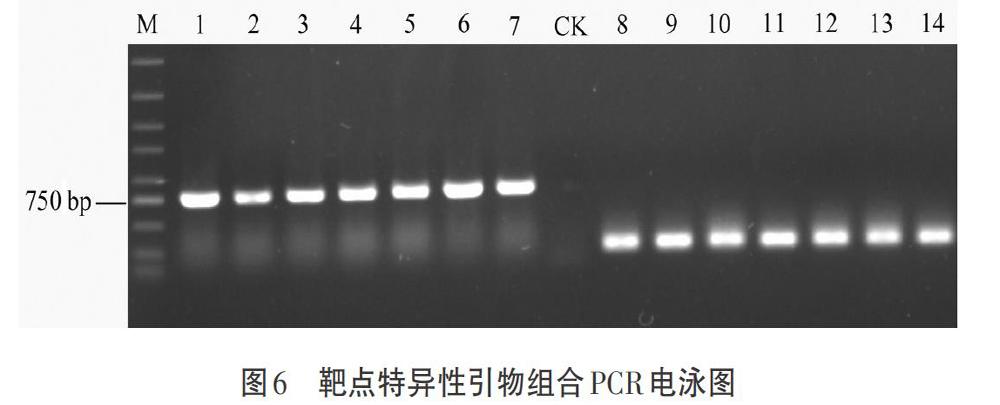
挑取部分陽性克隆用特異性引物再次進行PCR驗證,確定挑選菌的DNA鏈克隆插入方向(見圖6)。將構建好的載體菌液送出測序,測序同樣使用引物CRISPRCX-F和CRISPRCX-R,測序結果顯示完全正確,多次檢測表明載體pYLCRISPR/Cas9-gRNA-OsPE構建是成功的。將通過檢測的pYLCRISPR/Cas9-gRNA載體質粒導入感受態的農桿菌EHA105中,獲得的陽性克隆農桿菌即可用于侵染水稻愈傷材料。
3 討論
CRISPR/Cas9載體系統作為一種新興的基因編輯系統,在植物中已經有多例應用和驗證,目前尚未有利用CRISPR/Cas9載體系統對多胚相關基因編輯的報道。CRISPR/Cas9基因編輯系統相對于其他編輯技術效率更加顯著,且操作更便捷。相對于其他編輯技術,CRISPR/Cas9的編輯效率有了明顯的提升,容錯率也大大提高,且CRISPR/Cas9系統編輯不攜帶任何外源DNA,這是其他編輯方式所不具備的。
CRISPR-Cas9系統可編輯的位點在整個基因組范圍內分布頻率較高,很容易篩選出合適的靶點進行基因編輯。CRISPR-Cas9可以同時對基因組進行多位點編輯,大大提高了基因編輯的效率。該編輯系統的優勢還在于其載體構建簡單且靶向效率很高,只需要構建一個CRISPR sgRNA即可與DNA序列進行匹配,從而介導Cas9蛋白對DNA序列進行切割,同時可以更有效地去除假陽性。CRISPR/Cas系統是一種有效和通用的生物技術工具,將對基因組工程的未來發展產生重大影響[12]。
CRISPR的最終目的就是對其現有的基因組序列進行一系列編輯,通過外源T-DNA的插入,使水稻基因序列發生一定的突變,影響OsPE相關功能蛋白的合成和特定靶序列的識別,影響水稻外在的表型,最終得到突變的水稻植株,其后代種子發芽時可能出現兩個或者多個胚。
本研究通過CRISPR/Cas9系統,先借助靶點特異性引物,經過兩輪Overlapping PCR對U6啟動子進行擴增,得到一個相應sgRNA的表達盒,然后用Golden gate cloning法將sgRNA表達框克隆到載體中相應的靶位點,經過多次檢測與驗證,最終獲得了針對水稻多胚基因OsPE的pYLCRISPR/Cas9-gRNA編輯載體,為進一步利用基因編輯技術改良多胚基因的遺傳育種奠定基礎。
參考文獻:
[1] 劉向東,陳啟鋒,李維明.作物多胚現象研究綜述[J].福建農學院學報,1992,21(02):147-156.
[2] Puri A, Basha P O, Kumar M, et al. The polyembryo gene (OsPE) in rice[J]. Functional & Integrative Genomics, 2010, 10(03): 359-366.
[3] Zhang F, Wen Y, Guo X. CRISPR/Cas9 for genome editing: progress, implications and challenges[J]. Human Molecular Genetics, 2014, 23(01): 40-46.
[4] 張喻,江海霞,閆文亮,等.CRISPR-Cas9系統敲除亞麻FAD2基因表達載體的構建[J].分子植物育種,2019,17(07):2185-2192.
[5] Hryhorowicz M, Lipiński D, Zeyland J, et al. CRISPR/Cas9 immune system as a tool for genome engineering[J]. Archivum Immunologiae et Therapiae Experimentalis, 2017, 65(03): 233-240.
[6] Püllmann P, Ulpinnis C, Marillonnet S, et al. Golden Mutagenesis: An efficient multi-site-saturation mutagenesis approach by Golden Gate cloning with automated primer design[J]. Scientific Reports, 2019, 9(01): 10932.
[7] Chiasson D, Giménez-Oya V, Bircheneder M, et al. A unified multi-kingdom Golden Gate cloning platform[J]. Scientific Reports, 2019, 9(01): 10131.
[8] Razzaq A, Saleem F, Kanwal M, et al. Modern trends in plant genome editing: An inclusive review of the CRISPR/Cas9 toolbox[J]. International Journal of Molecular Sciences, 2019, 20(16): 4045.
[9] Ma X, Zhang Q, Zhu Q, et al. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants[J]. Molecular Plant, 2015, 8(08): 1274-1284.
[10] 張揚,黃維峰,周菲,等.1種簡單快速高效的雙靶點CRISPR/Cas9載體構建方法[J].華中農業大學學報,2020,39(03):9-18.
[11] 胡春華,鄧貴明,孫曉玄,等.香蕉CRISPR/Cas9基因編輯技術體系的建立[J].中國農業科學,2017,50(07):1294-1301.
[12] Sampson T R, Weiss D S. Exploiting CRISPR/Cas systems for biotechnology[J]. Bioessays, 2014, 36(01): 34-38.
(責任編輯:丁志祥)
