基于HPLC多組分定量測定結合化學計量學的慢支固本顆粒質量評價
2021-03-12 05:33:00姚建華王婧寧趙志國
實用藥物與臨床 2021年12期
姚建華,趙 博,王婧寧,趙志國,韋 杏,黃 群*
0 引言
慢支固本顆粒由黃芪、防風、白術和當歸組方而成,主要用于慢性支氣管炎緩解期因肺脾氣虛而出現的乏力、自汗、惡風寒、咳嗽、咯痰、易感冒、食欲不振等病癥的治療,對呼吸道細菌感染、支氣管哮喘具有一定的治療作用[1]。慢支固本顆粒現收載于中國藥典2020年版一部[1],質量標準和文獻報道僅對其所含的1~2個成分進行了定量控制,不能全面地反映制劑的整體質量[2]。為更好地保障臨床用藥的穩定性和療效一致性,本研究依據中醫君、臣、佐、使配伍原則,結合中藥質量標志物確認原則,采用HPLC法對慢支固本顆粒中8種主要成分的含量同時進行測定,以期建立多指標的慢支固本顆粒控制方法,同時采用SPSS 26.0統計軟件對定量結果進行聚類分析和主成分分析,為全面評價慢支固本顆粒整體質量提供科學數據。
1 儀器與試劑
1.1 儀器 1100型高效液相色譜儀(美國Agilent公司);KQ-250DB型超聲波清洗器(功率250 W,頻率40 kHz,昆山市超聲儀器有限公司);XS105DU型電子分析天平(精度:0.01 mg,德國Mettler Toledo公司)。
1.2 試劑 對照品毛蕊異黃酮葡萄糖苷(批號111920-201907,質量分數96.8%,CAS號20633-67-4)、升麻素苷(批號111522-201913,質量分數94.6%,CAS號80681-45-4)、白術內酯Ⅲ(批號111978-201501,質量分數99.9%,CAS號73030-71-4)、白術內酯Ⅰ(批號111975-201501,質量分數99.9%,CAS號73069-13-3),購于中國食品藥品檢定研究院;對照品9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷(批號PRF8101143,質量分數99.1%,CAS號94367-42-7)、芒柄花苷(批號PRF8081121,質量分數99.6%,CAS號486-62-4)、毛蕊異黃酮(批號PRF8062601,質量分數99.6%,CAS號20575-57-9)、升麻素(批號PRF9092523,質量分數99.7%,CAS號37921-38-3),購于成都普瑞法科技開發有限公司。慢支固本顆粒(規格:每袋裝10 g;批號:20190203、20190207、20190211、20190215、20190302、20190501、20190503、20190504、20190506和20190507,編號分別為S1~S10)來源于浙江新光藥業股份有限公司;乙腈為色譜純,其他試劑為分析純。
2 方法與結果
2.1 溶液的制備
2.1.1 混合對照品溶液的制備 精密稱取毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、毛蕊異黃酮、升麻素苷、升麻素、白術內酯Ⅲ、白術內酯Ⅰ對照品各適量,用甲醇制成質量濃度分別為0.498、0.264、0.372、0.196、1.498、0.534、0.158和0.112 mg/ml的混合對照品儲備液。精密吸取上述溶液0.1、0.5、1.0、1.5、2.0、2.5 ml,分別用甲醇定容至20 ml,制成6個系列質量濃度線性考察用混合對照品溶液;取中間質量濃度混對溶液作為混合對照品溶液(毛蕊異黃酮葡萄糖苷24.9 μg/ml、芒柄花苷13.2 μg/ml、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷18.6 μg/ml、毛蕊異黃酮9.8 μg/ml、升麻素苷74.9 μg/ml、升麻素26.7 μg/ml、白術內酯Ⅲ 7.9 μg/ml、白術內酯Ⅰ 5.6 μg/ml)。
2.1.2 慢支固本顆粒供試品溶液 取慢支固本顆粒樣品適量,剪開內包裝,傾出內容物研細,取1.0 g,精密稱定,置25 ml量瓶中,加入甲醇適量,超聲提取30 min,放冷后用甲醇定容,搖勻,濾過,即得。
2.1.3 陰性樣品溶液 按慢支固本顆粒質量標準項下處方工藝,分別制備缺黃芪、缺防風、缺白術的陰性樣品,再按上述方法制成3種陰性樣品溶液。
2.2 色譜條件及專屬性試驗 采用Agilent SB C18液相色譜柱(250 mm×4.6 mm,5 μm),流動相為乙腈(A)-0.2%磷酸溶液(B),梯度洗脫,見表1;流速為0.9 ml/min;檢測波長:254 nm(0~35 min檢測毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、毛蕊異黃酮、升麻素苷和升麻素)[3-8]和220 nm(35~55 min檢測白術內酯Ⅲ和白術內酯Ⅰ)[9-12];柱溫為30 ℃;進樣量為10 μl。精密吸取混合對照品溶液、供試品溶液和陰性樣品溶液各10 μl,進樣檢測并記錄色譜圖(圖1)。結果理論板數按各成分色譜峰計均>4 000,8種成分色譜峰分離效果良好,分離度均>1.5,陰性樣品對8種成分同時測定無干擾。
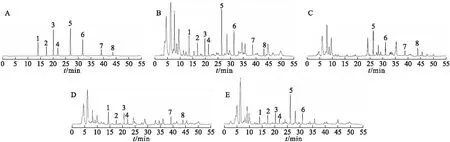
圖1 HPLC色譜圖

表1 梯度洗脫程序
2.3 線性關系考察 精密吸取“2.1.1”項下6個系列質量濃度線性考察混合對照品溶液適量,依次進樣測定慢支固本顆粒中8種目標化合物的峰面積,以各成分質量濃度(X,μg/ml)為橫坐標,峰面積(Y)為縱坐標,得8種目標化合物的回歸方程、r值及線性范圍,見表2。
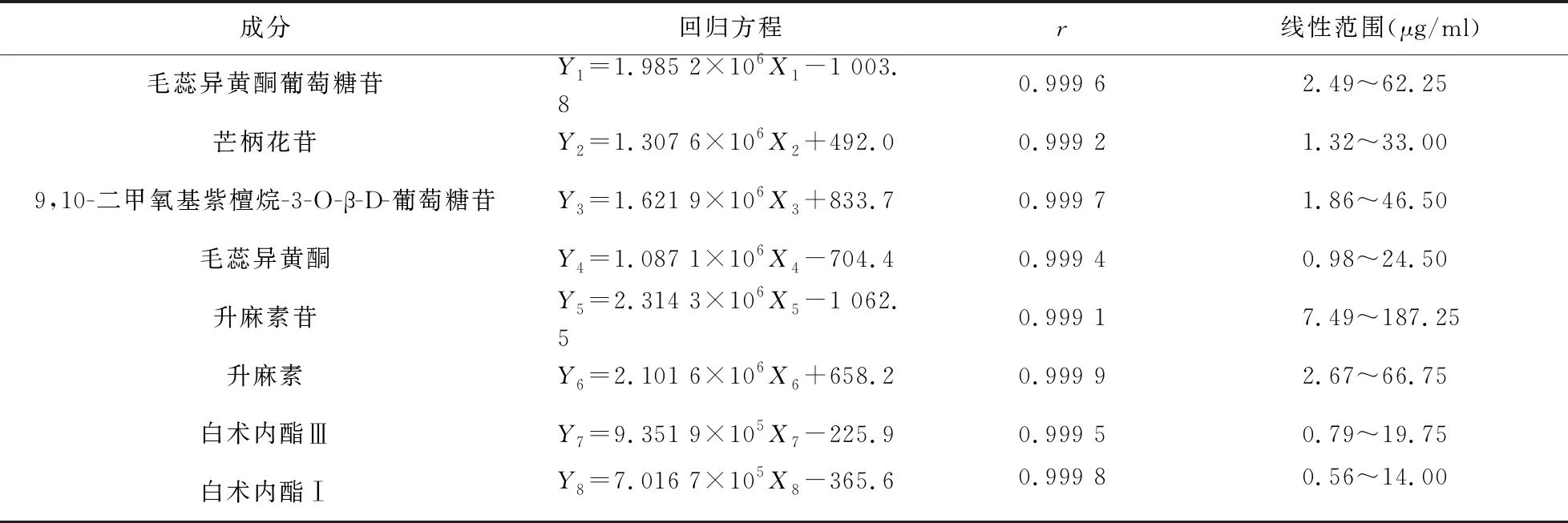
表2 慢支固本顆粒中8種成分線性關系
2.4 精密度試驗 取同一份慢支固本顆粒供試品溶液連續進樣6次,記錄毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、毛蕊異黃酮、升麻素苷、升麻素、白術內酯Ⅲ、白術內酯Ⅰ的峰面積,結果8種目標化合物峰面積的RSD分別為0.88%、1.01%、0.95%、1.14%、0.52%、0.79%、1.06%和1.23%。
2.5 重復性試驗 取同一批慢支固本顆粒樣品,平行制備6份供試品溶液,依法進樣檢測毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、毛蕊異黃酮、升麻素苷、升麻素、白術內酯Ⅲ、白術內酯Ⅰ的峰面積并計算各成分含量,結果8種成分含量的RSD分別為1.45%、1.62%、1.30%、1.51%、0.89%、1.08%、1.73%和1.86%。
2.6 穩定性試驗 取同一份慢支固本顆粒供試品溶液于0、2、4、9、18和24 h進樣檢測毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、毛蕊異黃酮、升麻素苷、升麻素、白術內酯Ⅲ、白術內酯Ⅰ的峰面積,結果顯示,慢支固本顆粒供試品溶液24 h內穩定,供試品溶液中8種目標化合物峰面積的RSD分別為0.90%、1.03%、1.01%、1.12%、0.58%、0.82%、1.11%和1.19%。
2.7 加樣回收率試驗 取8種成分含量均已知的同一批次慢支固本顆粒樣品適量,傾出內容物研細,取9份,每份0.5 g,精密稱定,精密加入根據8種成分含量另行配制的混合對照品溶液(毛蕊異黃酮葡萄糖苷0.326 mg/ml、芒柄花苷0.198 mg/ml、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷0.262 mg/ml、毛蕊異黃酮0.138 mg/ml、升麻素苷1.074 mg/ml、升麻素0.372 mg/ml、白術內酯Ⅲ 0.108 mg/ml、白術內酯Ⅰ 0.084 mg/ml)0.5、1.0、1.5 ml,再按供試品溶液制備方法制成加樣回收樣品溶液,依法進樣測定,計算各成分含量及加樣回收率,結果見表3。
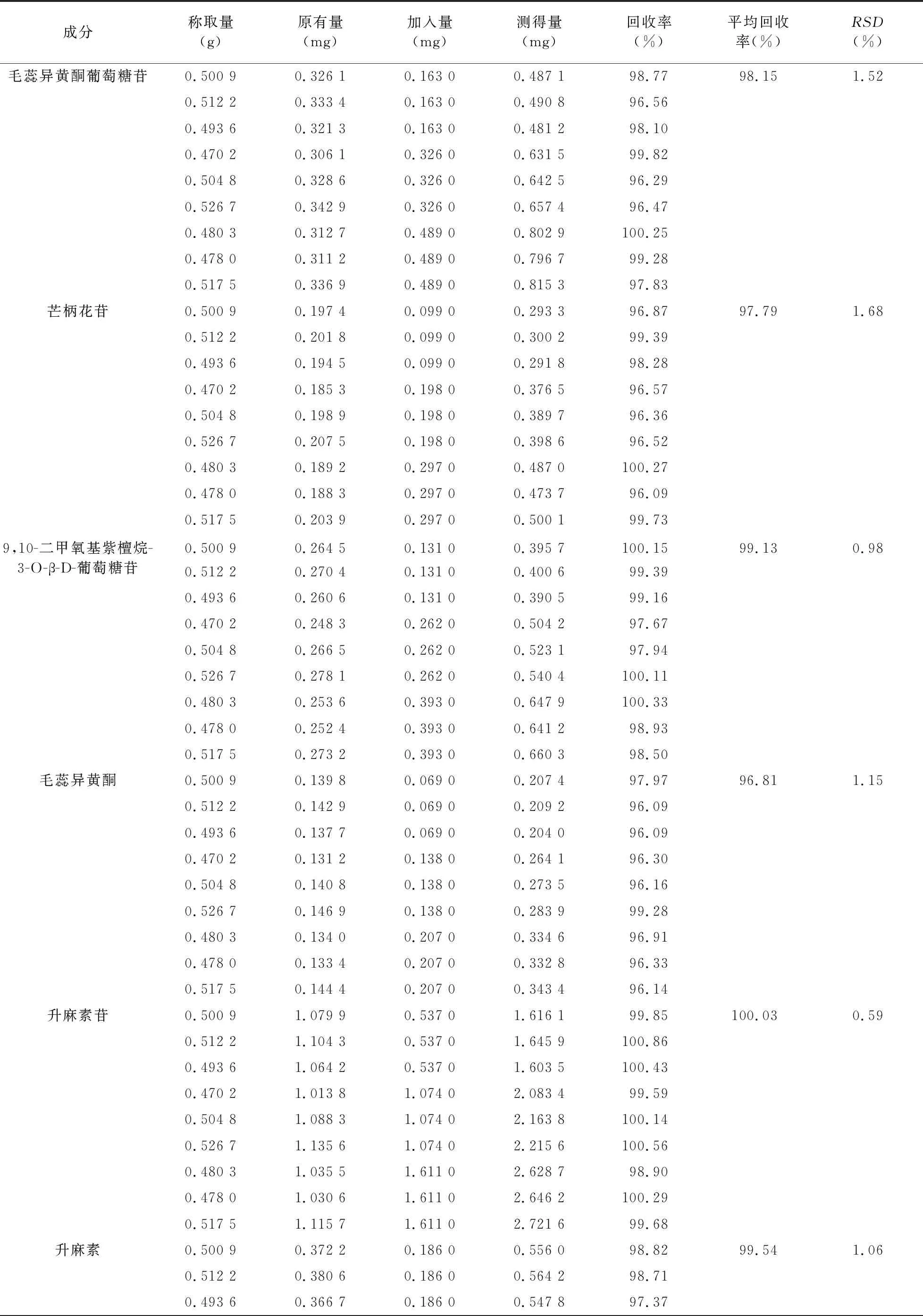
表3 慢支固本顆粒中8種成分的回收率實驗結果
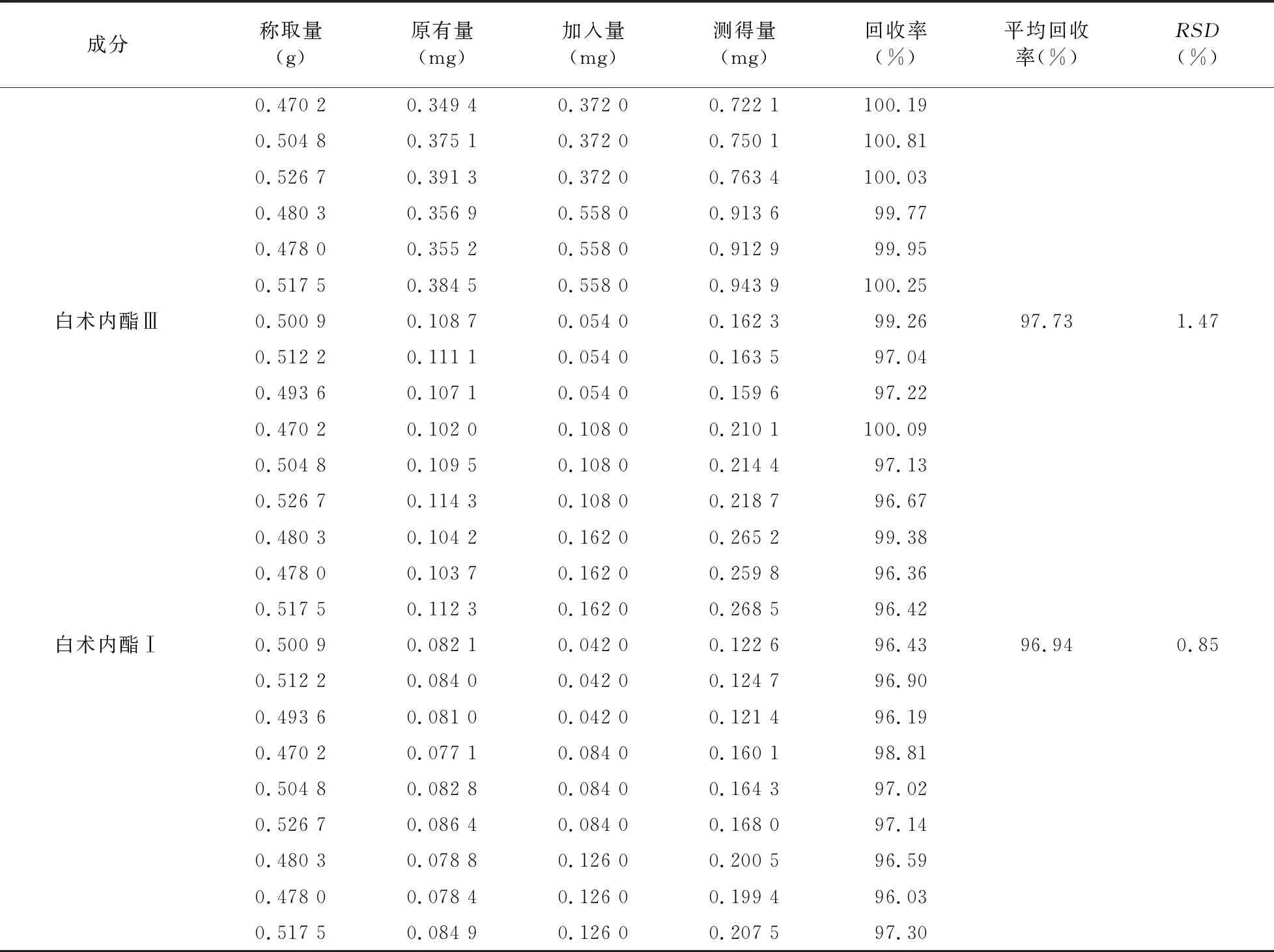
續表
2.8 樣品含量測定 取10個批次慢支固本顆粒樣品,按“2.1.2”項下方法制成供試品溶液,依法進樣檢測毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、毛蕊異黃酮、升麻素苷、升麻素、白術內酯Ⅲ、白術內酯Ⅰ的峰面積,計算8種目標化合物含量,結果見表4。
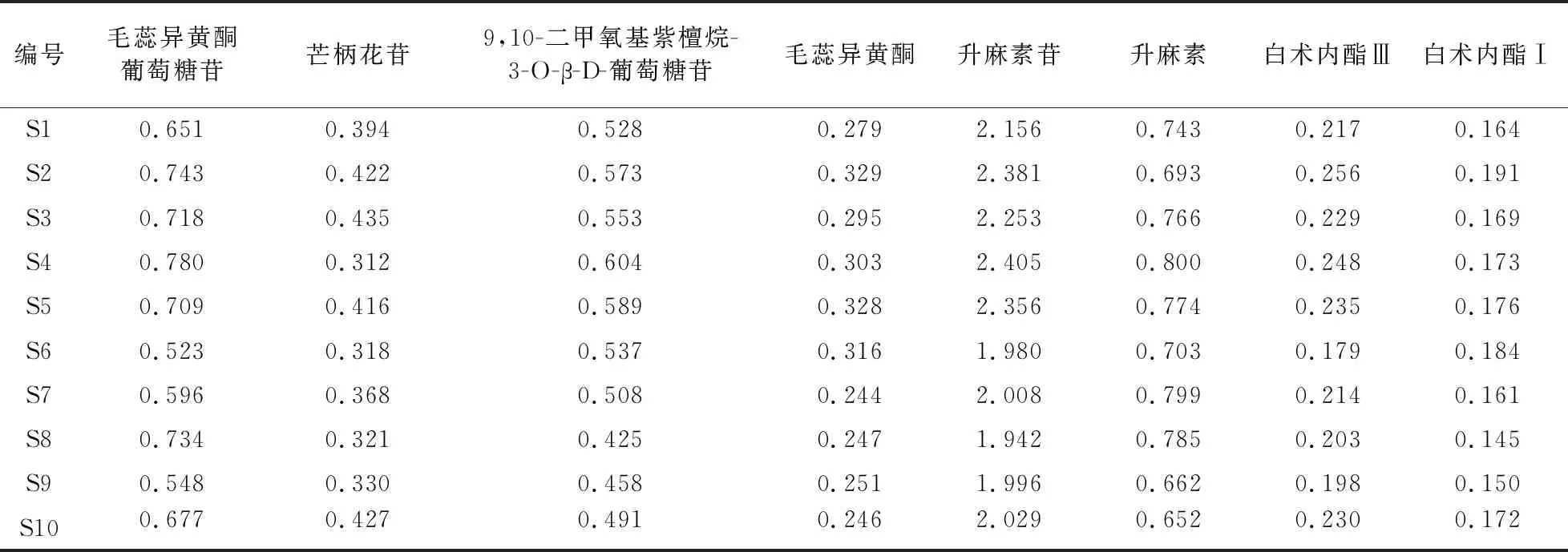
表4 樣品含量測定結果(mg/g)
2.9 化學計量學分析
2.9.1 聚類分析 為考察不同批次間慢支固本顆粒的含量差異,采用SPSS 26.0軟件組間聯接聚類分析方法,以歐氏距離為測度,以8種目標化合物毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、毛蕊異黃酮、升麻素苷、升麻素、白術內酯Ⅲ、白術內酯Ⅰ含量為變量,對10個批次慢支固本顆粒樣品進行聚類分析,結果見圖2。由圖2可知,當類間距離為15時,10批樣品聚為2類,樣品S2、S5、S4、S1和S3聚為第Ⅰ類,樣品S6、S9、S7、S10和S8聚為第Ⅱ類。從分類結果可以看出,目標化合物含量差異與樣品批次期間相關,可能與慢支固本顆粒生產所用原藥材的產地來源、采收時節不同以及原藥材的批間質量差異有關。
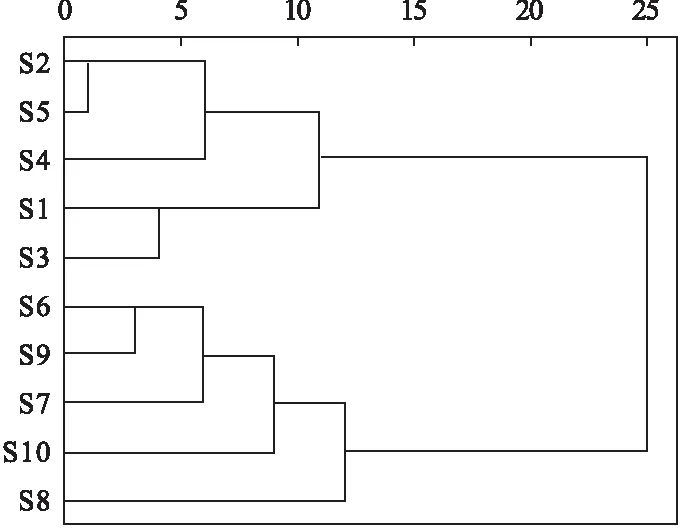
圖2 10批慢支固本顆粒聚類分析樹狀圖
2.9.2 主成分分析 采用SPSS 26.0軟件,以8種目標化合物含量為變量,對10個批次慢支固本顆粒含量數據進行主成分分析,得初始特征值和方差貢獻率見表5。

表5 主成分特征值和方差貢獻率
由表5可知,按照特征值>1,累積方差貢獻率>85%為提取標準,從慢支固本顆粒8種目標化合物中提取出前3個主成分進行分析評價。由表5可知,升麻素苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷、白術內酯Ⅲ、毛蕊異黃酮、白術內酯Ⅰ和毛蕊異黃酮葡萄糖苷是對主成分1影響較大的特征向量;升麻素對主成分2貢獻度高,芒柄花苷對主成分3貢獻度顯著。見表6。
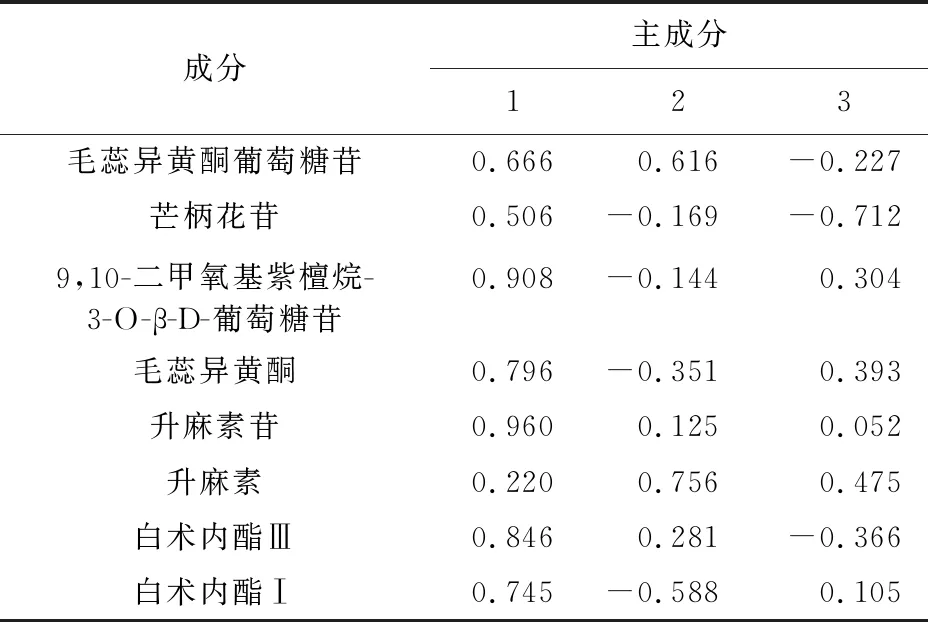
表6 10批慢支固本顆粒主成分分析結果
3 討論
3.1 定量目標成分的選擇 慢支固本顆粒由黃芪、防風、白術和當歸4味中藥材加工而成,方中黃芪補脾益肺、益氣固表,為君藥;白術健脾益氣、
利水止汗,助君藥黃芪補益脾肺,為臣藥;當歸補血和血,防風祛風解表、勝濕止痛,共為佐使藥,諸藥合用,共奏補肺健脾、固表和營之效。本文選取慢支固本顆粒君藥黃芪所含活性成分毛蕊異黃酮葡萄糖苷、芒柄花苷、9,10-二甲氧基紫檀烷-3-O-β-D-葡萄糖苷和毛蕊異黃酮,臣藥白術所含主要活性成分白術內酯Ⅲ和白術內酯Ⅰ,佐藥防風所含代表性成分升麻素苷、升麻素為定量測定目標成分,以期更加全面地評價慢支固本顆粒的整體質量。
3.2 流動相的優化 在流動相優化過程中,首先對不同有機相(甲醇、乙腈)進行對比分析,結果顯示甲醇-水體系[6-8]色譜圖基線不平穩,部分色譜峰拖尾嚴重,達不到定量檢測要求,而乙腈-水體系[11-13]基線稍平穩,但升麻素苷色譜峰出現拖尾現象。為有效改善峰形,采用乙腈為流動相中有機相,對不同水相(0.1%甲酸溶液[3-4,14-15]、0.1%磷酸溶液、0.05%磷酸溶液[10,16]、0.2%磷酸溶液)進行對比分析,同時對有機相和水相比例進行多次優化,最終選擇“2.4”項下的色譜條件。
中藥及其制劑,尤其是中成藥復方制劑相比于化學藥品,質量控制方法比較滯后,控制體系尚不完善,制劑生產所用原藥材因種屬來源、產地差異、生長年限、采收季節等因素影響,造成中藥制劑批間差異較大。從10批次樣品含量測定結果來看,8種目標化合物含量存在一定批間差異,編號S4、S5和S2樣品的8種目標化合物綜合含量較高,編號S9和S6樣品綜合含量較低,與聚類分析結果一致。本實驗采用HPLC法對慢支固本顆粒中8種成分含量同時進行測定,建立該制劑多指標成分質量控制模式,同時采用聚類分析、主成分分析等化學計量學手段對測定結果進行綜合評價,有助于藥品生產企業不斷完善制劑制備過程參數控制以及對原藥材內控質量標準的提升,為慢支固本顆粒質量控制體系的建立提供參考。中藥指紋圖譜具有信息量大、特征性強等特點,能夠呈現中成藥復方制劑所含更多化學成分信息,采用指紋圖譜結合化學計量學方法對該制劑進行綜合評價正在進一步研究中。
