支化GAP分子結構與流變性能表征研究①
2021-03-09 03:19:56安百強聶海英黃志萍
固體火箭技術 2021年1期
安百強,聶海英,黃志萍
(航天化學動力技術重點實驗室,湖北航天化學技術研究所,襄陽 441003)
0 引言
高分子聚合物的支化研究表明,長支鏈結構能顯著提升聚合物的熔體強度,提高熔體的剪切敏感度,有利于材料的加工成型[1]。提高聚合物的支化程度,如使其具有星型結構、超支化結構以及樹枝狀結構,可獲得良好的力學性能、流變性能以及表面多官能基團的特性[2]。如宋雪晶[3-4]使用超支化聚酯/超支化聚醚對以HTPB為基的聚氨酯形成互穿網絡,明顯改善了HTPB膠片的力學性能。
GAP是一種高能粘合劑,有著正生成熱、高密度、低感度和低玻璃化轉變溫度的優勢,與高能氧化劑有良好的相容性,在新型高能低特征信號推進劑中有良好應用前景[5-7]。線性GAP的羥基取代度低,應用線性GAP的疊氮聚醚類的推進劑力學性能較難提高。在數均分子量相同情況下,支化GAP比線性GAP燃燒產生更多的能量,粘度更低,玻璃化轉變溫度也有所降低[8],可望提高推進劑的流變性能和力學性能。目前關于支化GAP合成的文獻較多[9-11],對支化GAP支化程度、流變性能研究鮮有報道,這不利于支化GAP產品的質量控制。研究表征聚合物的支化度的常用方法有13C NMR法和激光光散射(LS)、粘度(CV)和示差折光(DRI)三檢測器與凝膠滲透色譜(GPC)聯用法。通過前期研究,支化GAP的13C NMR譜圖較為復雜,且支化點C原子數量較少,信號較弱,較難應用13C NMR法表征支化GAP支化度。近年來三檢測器與凝膠滲透色譜聯用技術在聚合物的支化度表征得到廣泛且較好的應用[12-14]。這是因為支化聚合物的流變性能不僅與支化度(分子形狀)相關,也受到分子量及分子量分布的影響[15],三檢測器正好可以對聚合物從分子量、分子形狀和流變性能三方面同時進行表征。例如聚乙烯樹脂的支化度和分子量對流變性能影響的研究[16-17]。
本文擬采用該技術對支化GAP的分子量、分子形狀等分子結構和流變性能進行表征研究,同時應用高級旋轉流變儀進一步表征支化GAP的動態流變性能,期望得到支化GAP分子結構與流變性能的關系,用于控制支化GAP產品質量,推廣其在新型推進劑中的應用。
1 實驗部分
1.1 試劑與儀器
1.1.1 試劑
支化GAP:編號BGAP-1、BGAP-2、BGAP-3,湖北航天化學技術研究所自制,重均相對分子量在100 000~200 000左右;線性GAP:編號GAP-1、GAP-2、GAP-3,湖北航天化學技術研究所自制,重均相對分子量在5000左右;四氫呋喃(THF):DIKMA,色譜純。
1.1.2 儀器
三檢測器與GPC聯用儀由以下部件組成:十八角度激光光散射儀(LS),美國Wyatt 公司DAWN HELEOS型;粘度檢測器(CV),美國Wyatt公司 ViscoStar-2型;示差折光儀(DRI),美國Wyatt公司 Optilab型;液相色譜單元泵,美國Agilent Technologies公司 1260 Infinity型;凝膠滲透色譜柱,日本Shodex公司 型號為KF-802.5、KF-803、KF-804的三根柱子依次串聯。
高級旋轉流變儀:美國TA公司ARES型。
1.2 實驗過程
1.2.1 流變性能測試
流變性能檢測:應用高級旋轉流變儀測試GAP樣品的頻率掃描動態粘彈曲線,流變儀的平板直徑為25 mm,板間距為1.5 mm,應變值為30%,溫度為70 ℃,頻率范圍為0.1~100 rad/s。
1.2.2 支化程度、分子量檢測
支化程度、分子量檢測:將不同批次的疊氮縮水聚醚分別用四氫呋喃溶劑溶解,支化GAP配制濃度為5 mg/ml,線性GAP配制濃度為20 mg/ml,搖勻靜置,放置超過8 h備用。
在測試溫度為40 ℃,流速為1 ml/min的條件下,進樣100 μl。樣品依次流過色譜柱、十八角度激光光散射儀(LS)、粘度檢測器(CV)和示差折光儀(DRI),繪制其三檢測器色譜變化曲線。
2 結果與討論
2.1 流變性能
2.1.1 動態流變性能
圖1為頻率掃描模式下支化GAP在70 ℃下模量與粘度隨頻率變化曲線。
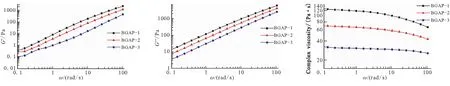
(a)Storage modulus G′ (b)Loss modulus G″ (c)Complex viscosity
由圖1(a)、(b)可知,三批支化GAP的儲能模量G′、損耗模量G″隨角頻率ω的增加呈線性增長,三者線性斜率無明顯區別,變化趨勢相近,大小隨樣品BGAP-1、BGAP-2和BGAP-3依次下降。而由圖1(c)可知,復合粘度則隨樣品 BGAP-1、BGAP-2和BGAP-3依次下降,且在角頻率為0.1 rad/s時,復合粘度較高,分別為131.7、69.7、33.5 Pa·s,而在角頻率為100 rad/s時,其復合粘度分別下降至65.7、43.2、26.0 Pa·s,下降幅度依次增加,分別為各自低頻率的49.9%、62.0%和77.6%。可見模量越大,粘度越大,且隨著角頻率增大粘度下降,各支化GAP具有剪切變稀行為。
2.1.2 零剪切粘度
高分子在溶液中或熔融狀態時如同糾纏的線球,分子鏈間的糾纏點維系著分子鏈間結構的穩定。零剪切粘度η0是剪切速率為零時的粘度,其大小可反應出聚合物的結構強度,實驗上無法獲得,可利用Cross模型對復合粘數η*與角頻率ω的曲線進行擬合得到零剪切粘度η0[18]。
(1)
式中λ為松弛時間;n為剪切變稀指數參數。
三批支化GAP的零剪切粘度數據如表1所示,可見三批支化GAP的零剪切粘度依次減小,BGAP-1的零剪切粘度是BGAP-3的4.72倍,是BGAP-2的1.75倍,說明BGAP-1的分子鏈間纏結作用最強,受到分子鏈間相互作用力最大,這與其動態模量和復合粘度規律一致。
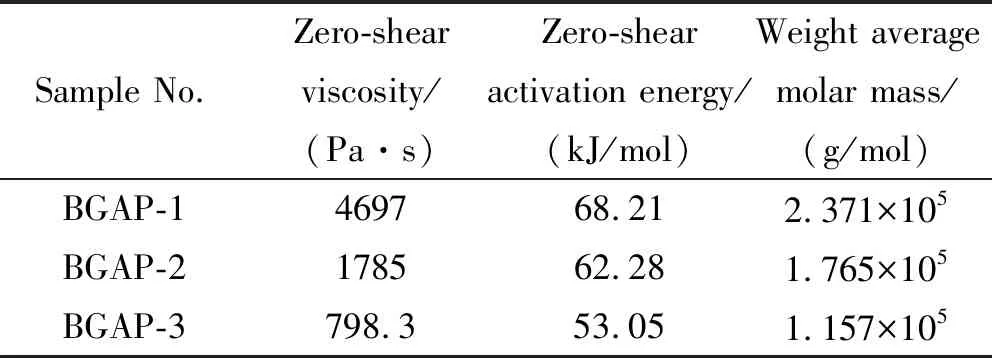
表1 支化GAP的零剪切粘度、零剪切活化能 和重均相對分子量
2.1.3 粘流活化能
高分子運動并不是整個分子的遷移,而是鏈段的相繼躍遷。在溫度升高后,分子鏈段的活動能力更強,克服鏈段間的摩擦能力變強,從而導致鏈段沿剪切應力方向取向更容易,分子鏈段運動所需克服的勢壘更小。粘流活化能可反應聚合物的粘度隨溫度變化的敏感性,粘流活化能越高,材料因溫度變化導致性能變化越大。支化GAP的粘流活化能可根據Arrhenius經驗式(2)計算:
(2)
式中η為粘度;A為常數;ΔEη為粘流活化能;R為氣體常數;T為絕對溫度。
改變溫度條件,在30、40、50、60、70 ℃下求得各支化GAP的零剪切粘度η0,根據式(2)用lnη0與1/T作圖如圖2所示,所得直線的斜率即為ΔEη0,零剪切活化能結果如表1所示。
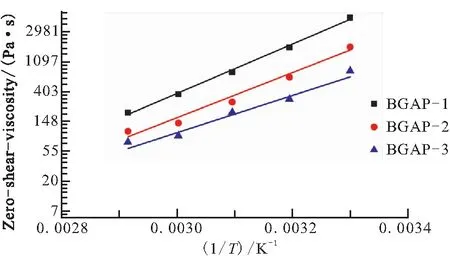
圖2 lnη0與1/T的關系圖
由表1可知,不同批次支化GAP的零剪切活化能不同,故其粘度對溫度依賴性不同,BGAP-1對溫度變化更敏感。各支化GAP對溫度變化敏感性隨樣品BGAP-1、 BGAP-2、BGAP-3依次下降。溫度由30 ℃升高到70 ℃后,BGAP-1、BGAP-2和BGAP-3的零剪切粘度分別大幅度下降到各自初始值的4.23%、6.00%和9.31%,升高溫度對鏈段間的解纏結作用非常明顯。因分子鏈解纏結運動是不同鏈段的行為[19],在外部的鏈段先解纏結松弛,隨后內部主鏈再運動松弛,故在高分子聚合物中零剪切活化能不僅與分子鏈段長度(相對分子質量)相關,也與其支化程度相關。
2.2 重均相對分子量
三檢測器與GPC聯用可直接得到聚合物的絕對重均相對分子量,不需要標樣校準。應用激光光散射檢測支化GAP相對分子質量結果如表1所示。不同批次的支化GAP間分子量差異較大,BGAP-1的Mw是BGAP-3的2.05倍,是BGAP-2的1.34倍。由此可知,重均相對分子量增大使得分子鏈間的相互作用因纏結而增強,導致零剪切粘度明顯增大。若假設三批支化GAP的Mw均在臨界相對分子質量Mc以下,則零剪切粘度與重均相對分子量的關系可用式(3)表示:
(3)
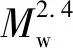
2.3 支化程度
盡管利用三檢測器與GPC聯用技術檢測聚合物的支化度技術已經非常成熟,并得到廣泛應用,但支化GAP沒有與之相同重均相對分子量的線性GAP,使得支化GAP的支化程度檢測仍為有待解決的問題。本文選取了低相對分子質量線性GAP和分布較寬、重均相對分子量為200萬的線性聚苯乙烯PS分別作為線性標樣,用三檢測器與GPC聯用法檢測支化GAP支化程度,分別用特性粘數法與均方回轉半徑法計算,以確定其結果的可靠性。
2.3.1 特性粘數法
特性粘數定義支化因子g′[21],如式(4)所示:
(4)
其中,[η]br與[η]lin為相同相對分子質量下的支化分子特性粘數與線性分子特性粘數。在相同相對分子質量的支化聚合物,因支化程度不同,有不同數量的長鏈或短鏈,導致線團緊縮,在溶液中特性粘數會比線性聚合物的特性粘數有所降低[22],g′值通常在0~1之間。
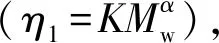
GAP的的K和α值結果如表2所示。
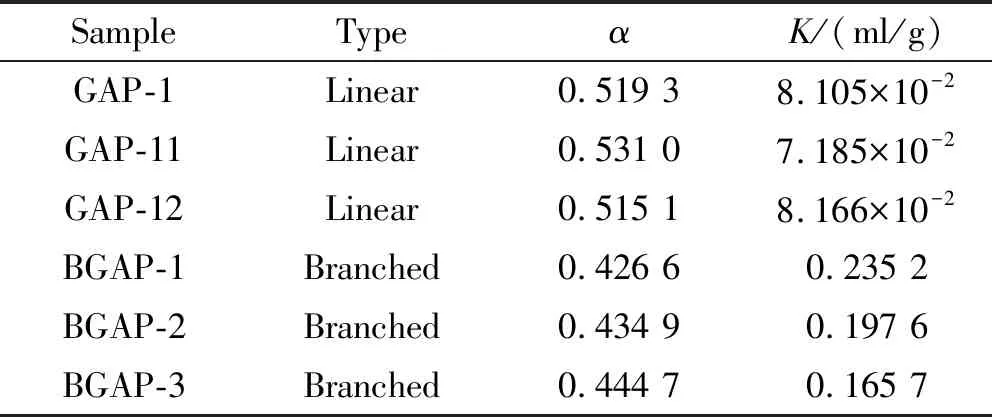
表2 GAP的K、α測定結果
不同線性GAP與支化GAP的特性粘數與相對分子質量關系如圖3所示。由表2的數據可知,線性GAP的α值在0.51~0.53左右,而支化GAP的α值在0.42~0.45左右,支化GAP的線團更為緊密。由圖3可知,支化GAP在重均相對分子量超過100 000后,特性粘數明顯小于線性GAP的特性粘數,支化GAP的線團更為緊密,符合支化聚合物的特性粘度表現,故利用線性GAP的K和α值擬合出高相對分子質量線性GAP特征粘數用于計算支化GAP支化因子是可行的。
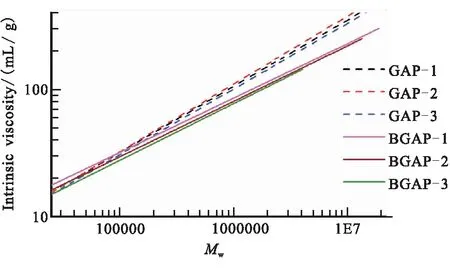
圖3 GAP的特性粘數與重均相對分子量關系
GAP-1、GAP-2與GAP-3為小分子線性GAP,分別以3個線性GAP作為線性標樣,利用公式計算3個支化GAP的支化因子,結果如表3所示。
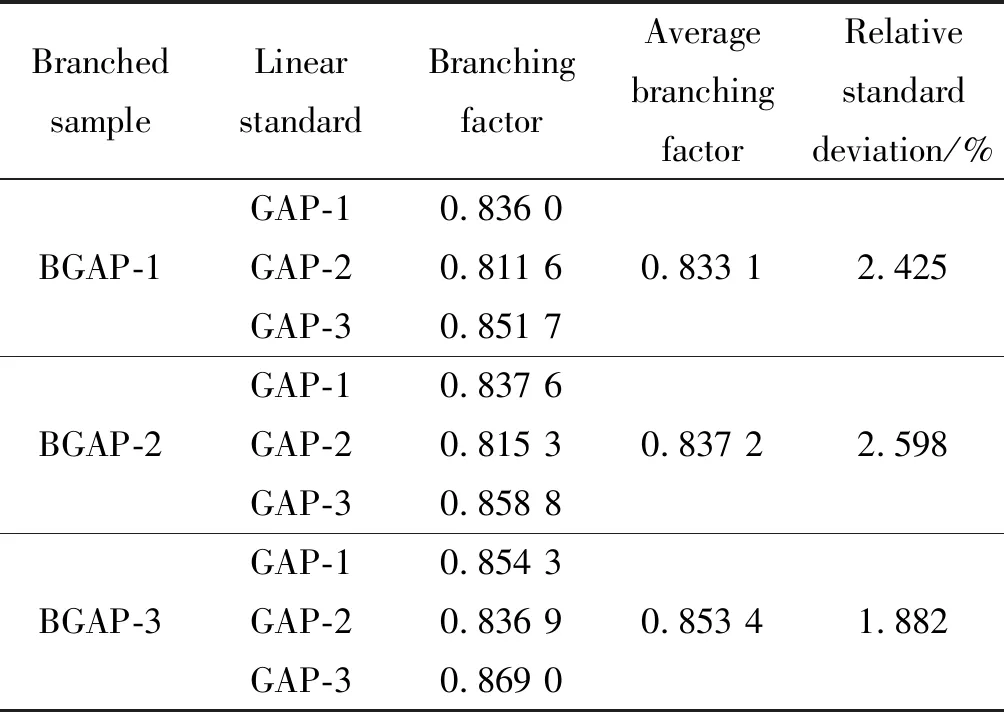
表3 特性粘數法計算支化GAP的支化因子
從表3可知,雖然選擇不同線性分子,計算支化GAP支化因子的結果略有不同,但各批次支化GAP的支化程度大小順序相同,且參照不同線性分子所得支化因子結果的相對標準偏差小于3%,說明不同批次線性GAP的影響較小,方法是可行的。所得支化GAP之間總支化程度的大小隨樣品BGAP-1 、BGAP-2、BGAP-3依次下降,但下降程度較小,故不同批次支化GAP的支化程度對模量和粘度的降低作用不明顯,重均相對分子量差別較大,對模量和粘度的增強作用更明顯。
2.3.2 均方回轉半徑法
均方回轉半徑法計算支化GAP的支化程度是利用三檢測器與GPC聯用儀檢測支化聚合物和線性聚合物的回轉半徑Rg,再用式(5)計算支化因子g[23]。在相同相對分子質量下,支化聚合物的均方根半徑(Rg br)小于線性聚合物的均方根半徑(Rg lin),g值也在0~1之間。
g=(Rg br/Rg lin)2
(5)
線性GAP的重均相對分子量較小(在10 000以下),其回轉半徑在儀器檢測下限10 nm以下,無法獲得線性GAP的回轉半徑Rg與重均相對分子量的關系。故選用了線性聚苯乙烯作為標樣,其廣泛易得,重復單體的相對分子質量為110,而GAP的重復單體的相對分子質量為99,兩者重復單體的相對分子質量較為相近,且PS和GAP有相似結構的側位取代基團,故在相同重均相對分子量下線性標樣的回轉半徑也較為相近。選用分布較寬、重均相對分子量為2 000 000的PS作為線性標樣,所得均方回轉半徑Rg與重均相對分子量Mw的關系如圖4所示。
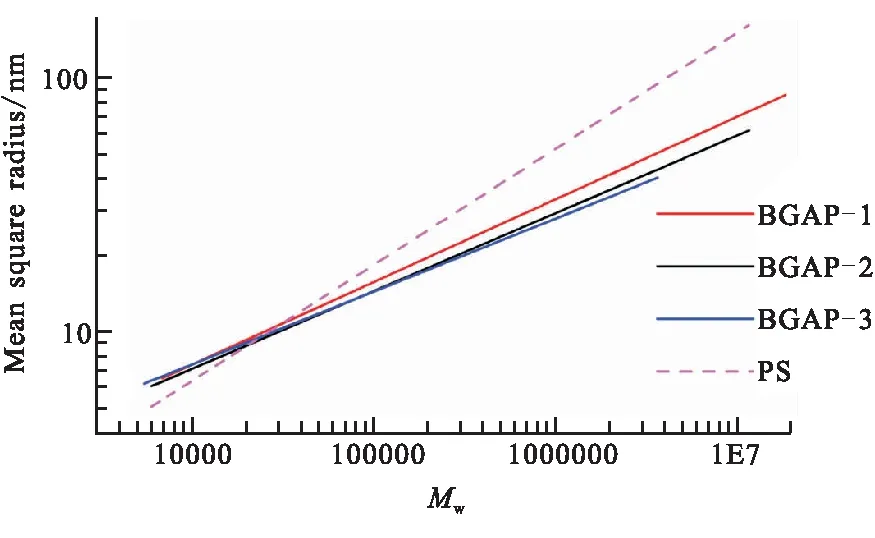
圖4 Rg與重均相對分子量Mw的關系圖
由圖4可知,在重均相對分子量30 000以上,PS的回轉半徑大于支化GAP的回轉半徑,可計算不同支化GAP的支化程度,表明選用線性聚苯乙烯作為線性標樣是可行的。而在小相對分子質量部分,PS的回轉半徑小于支化GAP的回轉半徑,說明小分子GAP未達到支化的臨界相對分子質量M*。
用式(5)計算不同支化GAP的支化因子,結果如表4所示。結果表明,均方回轉半徑法計算得到支化因子比特性粘數法支化因子小,而大小變化規律與特性粘數大小變化規律與特性粘數法一致。這是因為GAP重復單體的相對分子質量較PS小,PS的鏈段剛性較強,同樣相對分子質量的線性GAP的回轉半徑會略小于PS的回轉半徑,因此以PS作為線性標樣所計算的支化因子比實際偏小,即支化程度偏大。故在沒有較大相對分子質量GAP線性標樣的情況下,采用特性粘數法支化因子更接近實際結果。采用線性聚苯乙烯作為線性標樣檢測支化GAP的支化因子也是可選擇的方法。

表4 支化GAP的均方回轉半徑法支化因子
3 結論
(1)通過動態流變性能、零剪切粘度、粘度活化能,可表征不同支化GAP的流變性能差異。
(2)支化GAP有剪切變稀行為。
(3)特性粘數法與回轉半徑法測定支化GAP的支化程度方法可行,結果規律一致。在沒有較大相對分子質量GAP線性標樣的情況下,采用特性粘數法支化因子更接近實際結果。
(4)支化GAP的重均相對分子量和支化程度對流變性能有相關性,其差別程度越大,流變性能的差別越顯著。
