放射性污染土壤中銫的吸附/解吸行為研究進展
2021-03-06 05:27:22王善強李戰國趙紅杰韓夢薇
原子能科學技術 2021年3期
關鍵詞:模型
張 坤,王善強,李戰國,齊 圣,趙紅杰,韓夢薇
(1.國民核生化災害防護國家重點實驗室,北京 102205;2.國防工程研究院,河南 洛陽 471023)
核試驗、核事故、核設施退役、核技術應用等人類核活動向環境中釋放了大量的放射性核素,導致生態環境(空氣、土壤、水體、植被等)受到不同程度污染[1]。放射性銫(Cs)在鈾核裂變中產額大,是環境放射性污染的重要評價指標,也是人類活動釋放到陸地生態環境中最重要的放射性核素[2]。近年來,放射性Cs在陸地生態系統中的吸附、解吸、遷移、轉化、歸趨等環境化學行為受到了廣泛關注。
土壤和沉積物中的黏土礦物是土壤、沉積物、基巖等物質最重要的組成部分,是控制放射性Cs環境化學行為和歸趨的重要固態介質,黏土礦物是控制放射性Cs在土壤中吸附、解吸、擴散、遷移等環境化學行為的關鍵因素。上述環境化學行為又進一步受到環境中不同介質之間的物理、化學、生物作用影響。放射性核素的地球化學性質受到土壤中的膠體顆粒、腐植酸、金屬離子、微生物等多種因素影響。為全面了解和系統評估土壤中放射性Cs的環境化學行為,需對土壤中放射性Cs的吸附/解吸行為及其作用機制開展系統研究。本文擬在文獻調研的基礎上,對放射性Cs在黏土礦物中的吸附/解吸行為及其表征方法進行比較和總結,并就未來的發展進行展望,為放射性污染修復治理、核設施退役廢物地質處理與處置等研究提供參考。
1 放射性Cs的來源
自1945年以來,全世界共開展了2 000多次核試驗,核試驗造成全球范圍核污染擴散,對地球環境和生態系統造成長期危害[3]。土壤中放射性核素的性質和來源列于表1,放射性Cs主要有137Cs和134Cs兩種同位素。切爾諾貝利和福島核電站事故給生態環境造成極大危害[4],切爾諾貝利核事故造成4 300 km2范圍內的土壤受到137Cs、90Sr、154Eu、238Pu、241Am等多種放射性核素不同程度的污染,事故導致大量放射性物質擴散并影響其他國家,如白俄羅斯、俄羅斯、德國、奧地利、希臘等國家[5];日本福島核事故造成周邊1 778 km2陸地范圍內建筑物、道路、土地、森林植被污染,137Cs污染土壤總量達1 600萬~2 200萬m3[6]。2次事故后的監測結果表明,切爾諾貝利事故對環境的輻射后果影響遠超過福島事故,該核事故使全球區域的森林、土壤和河流、海域等受到不同程度的污染;日本福島核事故釋放的大多數放射性核素沉降于近海并沉積在太平洋。

表1 土壤中主要放射性核素性質與來源Table 1 Property and source of main radionuclide in soil
Cs是一種堿金屬,與K的化學性質類似。核能利用導致大量的放射性Cs同位素釋放于環境,特別是長壽命放射性核素137Cs(T1/2=30.2 a)[7]。137Cs通過β-衰變生成137Bam(T1/2=2.55 min),約占總β-衰變的95%,能量為0.51 MeV,隨后衰變為穩定的137Ba,約占總β-衰變的5%,能量為1.17 keV。137Cs在鈾核裂變中的產額大,生物利用度高,可通過生態系統和食物鏈危及人類健康。因此,137Cs污染已成為當今國際社會高度關注的問題。
2 黏土礦物結構與特性
常見的黏土礦物屬層狀構造硅酸鹽礦物,由硅氧四面體和鋁氧八面體構成[8-9]。黏土礦物可變負電荷層結構是其對陽離子吸附、解吸、生物有效性、擴散、傳輸的重要理論依據。永久負電荷或層間電荷通過同晶取代作用產生,主要來源有兩種方式:一是四面體片(T)中的Si4+被Al3+取代;二是八面體片(M)中的三價陽離子(Al3+、Fe3+)被Mg2+取代。通過此類同晶取代作用,T-M-T層間被更多可交換陽離子占據,這種負電荷結構是2∶1型層狀硅酸鹽黏土礦物的重要特性之一[10]。
理想的1∶1高嶺石(二八面體)中,Si4+被Al3+取代,則高嶺石的分子式為(Si3Al1)-Al4O10(OH)8,由于高嶺石外表面附近的堿性陽離子Na+和K+的平衡作用,高嶺石的凈電荷應為-1。但1∶1型層狀硅酸鹽的層電荷通常接近于0,這是由于層間陽離子交換作用所導致的。可變負電荷特性在2∶1型層狀硅酸鹽和云母類礦物中較為明顯,2∶1型蒙脫石和云母的負電荷范圍為0.2~1.0[11]。
此外,層間膨脹性是影響黏土礦物對放射性Cs吸附性能的另一重要因素,伊利石和綠泥石為非膨脹性礦物,蛭石為中等膨脹,蒙脫石具有較大的膨脹性,綠泥石層間位置被氫氧化物(Al(OH)3)所占據,蛭石和蒙脫石凈電荷分別為0.6~0.9、0.2~0.6,伊利石凈電荷最大(0.9~1.0),其層間的固定能力最強。黏土礦物的膨脹性有助于層間陽離子交換能力和放射性Cs的吸附親和力提高。如通過考察伊利石、蛭石、蒙脫石和綠泥石對Cs的吸附能力表明,其對Cs的吸附能力大小順序為:蛭石>伊利石>蒙脫石>綠泥石[12]。
3 放射性Cs在黏土礦物中的吸附/解吸行為與過程
3.1 吸附位點
黏土礦物吸附放射性Cs的位點類型與結合形態可分為三吸附位點和五吸附位點,如圖1所示。三吸附位點包括:基面位點、楔形位點和層間位點。基面位點對Cs+的親和力最弱,選擇性差;風化作用形成的楔形位點對低水合能的Cs+具有較強的親和性和較高的選擇性(相對于其他堿金屬和堿土金屬離子);層間位點由楔形位點的Cs+擴散形成,對Cs+的親和性和選擇性僅次于楔形位點(圖1a)[13-15]。五吸附位點包括基面位點、邊緣位點、層間位點、楔形位點和水合層間位點(圖1b)[16],邊緣位點吸附特征與基面位點相似,水化夾層的吸附可促進楔形位點吸附,Cs+在黏土礦物中的吸附主要通過離子交換反應實現[17]。
3.2 吸附作用機制
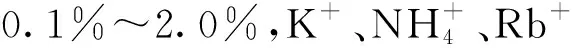

圖1 黏土礦物中Cs+吸附位點示意圖Fig.1 Schematic diagram of Cs+ adsorption site on clay mineral
Park等[20]提出了Cs+在伊利石上的吸附、遷移和非可逆性固定機制,如圖2所示。一種非可逆固定機制是Cs+使楔形位點塌陷(圖2a),Cs+選擇性吸附在楔形位點上,Cs+吸附后楔形位點塌陷,楔形位點結構與正常夾層或邊緣位點尺寸接近,Cs+遷移擴散至更深的層間區域;另一種作用機制是離子水合作用,由于K+的水合能高于Cs+,水合后的Cs+選擇性吸附在楔形位點上,楔形位點上的水合Cs+脫水,楔形位點附近中間層的K+被水合,從而使Cs+和K+的位置發生置換,脫水后的Cs+在中間層中最終變為非可逆固定(圖2b),這兩種作用機制可充分解釋Cs+在伊利石中與楔形位點和層間位點的選擇性吸附與非可逆性過程。
3.3 測定黏土礦物中楔形位點容量的方法
通常采用放射性Cs截留電位(RIP)法和銀硫脲(AgTU)法測定黏土礦物中楔形位點的容量。AgTU法是利用AgTu與黏土礦物基面位點結合并具有很強吸附親和力的特點,假設所有基面位點因與AgTu的絡合作用而被完全占據[18],Cs+只吸附在楔形位點上[21]。因此,利用過量AgTU可估算楔形位點吸附容量[22]。RIP法[23]與AgTU法原理相同,選用Ca2+和K+作為Cs離子取代陽離子。Wauters等[24]利用Ca2+和K+混合物進行了RIP測定,確定了楔形吸附位點含量。
AgTU法和RIP法主要通過分別添加高濃度的AgTU和Ca2+,使土壤黏土礦物的基面位點被AgTU和Ca2+吸附,而Cs+只能選擇性吸附于楔形位點,楔形位點吸附容量約占土壤黏土礦物中總CEC的0.25%~1.76%。AgTU法是土壤中特定位點數量選擇性吸附定量分析的技術基礎,與AgTU法相比,RIP法操作簡便且精確度高,更常用于估算黏土礦物中楔形位點的含量。
3.4 吸附模型
1) 等溫吸附模型
等溫吸附模型是基于平衡態和恒溫態下液相和固相核素濃度之間的數學關系,常用吸附模型有Langmuir模型、Freundlich模型、Langmuir-Freundlich(L-F)模型和Dubini-Radushkevich(D-R)模型[25]。
Langmuir模型假設基于平衡狀態下吸附和解吸分子數量在單位時間內相等,忽略礦物表面上的橫向吸附相互作用和水平遷移的影響,表面吸附位置分布均勻;Freundlich模型是描述非均勻相吸附體系的經驗模型,若固體表面不均勻,吸附平衡常數與表面特性有關;L-F模型是兩個模型的結合。

圖2 Cs+在伊利石上的選擇性吸附和遷移過程 Fig.2 Selective adsorption and migration of Cs+ on Illite
上述4個模型已用于研究放射性核素在黏土固液界面的吸附與解吸行為。Shahwan等[26]的研究表明,Cs在高嶺石、斜發沸石、膨潤土上的吸附與Freundlich和D-R模型吻合較好。花崗巖對Sr的吸附符合Langmuir模型,而花崗巖對Cs的吸附為非線性[27]。批式吸附實驗表明,紅壤對Cs+的吸附與Freundlieh等溫模型吻合較好[28]。
吸附等溫模型在金屬離子的吸附作用宏觀研究中應用很廣泛,實驗中不考慮酸堿度對吸附的影響,也不能描述吸附相表面結構信息、金屬離子在吸附相表面的化合態、表面吸附位點、累積濃度等適用范圍。因此,經驗吸附模型不能較好地解釋放射性核素與黏土礦物界面吸附與解吸的作用機理。
2) 表面配位模型
表面配位模型(SCM)是一種考慮表面電荷作用、吸附劑表面位點和吸附質分子的特異性、表面配合物分子的形態、質量作用規律、物質平衡規律等多種微觀和宏觀作用機制的描述方法[29]。
Schindler等[30]提出了配位化合物分子的描述方法,證明了陽離子在表面帶正電性吸附的可能性,表面配位模型既考慮了溶質在表面位點的內親和力作用,又關注了帶電表面與溶解離子之間的庫侖相互作用。
兩性表面水化羥基吸附是pH值影響的結果,而非土壤礦物溶解或H+交換[31],因此,表面配位作用機制可用于描述痕量Cs+在兩性羥基上的吸附。如,Gutierrez等[32]利用三層模型(TLM)模擬了Cs+在Ca基蒙脫石上的吸附,層間和楔形邊緣位點是Cs+吸附的主要原因,吸附的作用機理是邊緣部位的特異性吸附,邊緣吸附位點僅占總吸附位點的5%,但其吸附總量占比為94%。Silva等[33]提出了蒙脫石的另一種組合方法:(1) 具有1-pK雙層模型(DLM)的雙吸附位點(≡SOH和≡TOH基團);(2) 單位點陽離子交換模型,考慮了Cs+和Na+在≡SOH和≡TOH基團上的配位作用,該方法的缺點是需用12個參數進行擬合,應用過程復雜。Hurel等[34]利用一種表面配位模型研究了膨潤土對陽離子(Cs+、Na+、K+、Ca2+和Mg2+)的吸附,利用硅醇邊緣位點和陽離子交換模型模擬了Cs+的吸附過程。Wang等[35]還發現,在高pH值條件下,膨潤土對Cs+的吸附以表面配位作用為主。
表面配位模型中的靜電相互作用表現為界面區域的不同平面和層數,離子被吸附在不同的位置,常用于探討黏土礦物與Cs+的吸附機理。采用該模型可得到Cs+在土壤表面和溶液中的吸附形態,通過體系變化可得到相關的反應常數。然而,該模型適用于較理想的均質土壤類型,對于土壤這種典型固液非均相體系,在實驗上很難區分化學吸附能和靜電庫侖能兩種不同的相互作用。
3) 離子交換模型
離子交換理論是由Bolt(1982)提出的一種宏觀方法,該吸附理論涉及黏土礦物層間陽離子補償的結構負電荷。通常,可用多個交換位點來校準離子交換量,離子交換模型最初用陽離子交換容量的單點模型來描述,隨著研究的深入,逐步發展為多點位離子交換模型[18]。
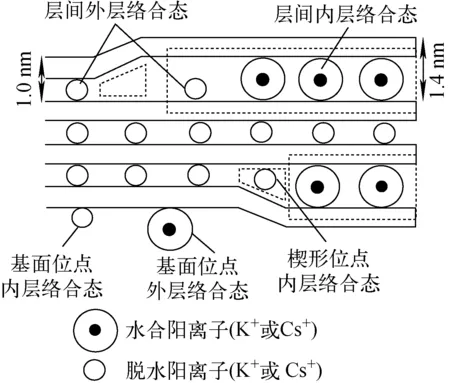
圖3 云母礦物吸附形態和吸附位點示意圖Fig.3 Adsorption morphology and site of mica minerals
研究表明,在大多數土壤中,Cs+離子主要吸附在伊利石楔形位點上(圖3)[36-37]。由于Cs+幾乎完全以一價陽離子的形式存在于溶液中,表面配位模型很難對Cs+的行為做出準確預測,Freundlich吸附模型說明Cs+在伊利石上至少有2個不同的吸附位點[38]。為模擬實驗數據,了解黏土礦物對Cs的非線性吸附行為,文獻中提出了一系列多點離子交換模型。對于伊利石、蒙脫石和高嶺石通常考慮三位點的陽離子交換模型[39-41]。此外,單位點的陽離子交換模型[42-43]和雙位點吸附模型[44]也在描述蒙脫石中Cs+的吸附行為研究中得到應用。
Bradbury等[40]建立了一種土壤與Cs的廣義吸附模型(GAM),該模型已用于分析不同的實驗結果。GAM的基本假設為:(1) Cs+在黏土礦物中的吸附機理為陽離子交換。(2) 存在3種不同的吸附位點類型,它們具有不同吸附容量和親和性,包括FES、PS和TIIS(由表面Si—O—Si和Al—O—Al斷裂形成的邊位點)。PS位點濃度與云母類礦物的CEC直接相關(如伊利石為固化體時,其PS位點約占CEC的80%),且吸附形態主要為外層絡合態(OSC);TIIS位點濃度低于PS(如伊利石為固化體時,TIIS位點約占CEC的20%)。由層狀云母類礦物風化形成的FES位點,其位點濃度相對較小(<10-6mol/g),但其對Cs+的選擇性吸附能力遠高于PS和TIIS位點,易形成內層絡合態(ISC)。對于Cs+,非膨脹性性云母礦物是IS位點,不可利用,而膨脹性云母類礦物可提供高容量的IS位點,當水合Cs+進入IS位點后失去水合氛圍,進而誘導層間坍塌至1 nm,此時,位于坍塌層間的Cs+主要以ISC形式存在,活性很低,形成了熱力學上更加穩定的結構。
在經典吸附模型的基礎上,Fan等[37]提出了Cs在蛭石上吸附行為的四位點優化模型,增加了IS位點的貢獻并優化了選擇性系數,得到各位點容量、選擇性系數和位點百分比參數,驗證了GAM改進模型在準確預測沉積物中Cs+分布的可能性。吳涵玉[45]利用四位點的吸附模型,研究了典型云母礦物與放射性Cs的吸附作用機制,連續提取實驗和EXAFS光譜澄清了Cs+在風化后云母礦物上的吸附形態,放射性Cs和Th(Ⅳ)/Eu(Ⅲ)復雜體系共存時,2類離子對金云母層間距離的調控具有拮抗作用,2類離子的相對濃度決定云母礦物的層間距離,從而控制云母礦物的風化過程,放射性Cs、Th/Eu調控金云母風化的機理如圖4所示。
綜上所述,單位點、雙位點、三位點、四位點、五位點等離子交換模型已在不同的黏土礦物吸附與解吸行為研究中得到應用。現有離子交換模型與一些實驗結果吻合較好,但模型中擬合參數多且不易確定。此外,模型僅適用于校準后的實驗數據,在不同土壤環境或自然系統中預測放射性Cs吸附、解吸行為的能力有限。環境中土壤結構類型復雜,單一的離子交換模型難以滿足應用需求,因此,需結合環境中土壤類型,開展多種吸附模型融合研究。
4 吸附/解吸行為表征方式及參數
Cs+在黏土礦物中的吸附、解吸作用機制的研究通常是利用光譜法和顯微分析技術進行量化分析。
4.1 核磁共振譜分析
核磁共振(NMR)譜的工作原理是原子在磁場中會與特定頻率的電磁波發生共振,NMR譜儀通過射頻共振測量樣品中原子的自旋[46]。NMR譜可用于13C、1H、39K[47]、7Li[47]、23Na[48]、133Cs[49]等陽離子元素分析。其中,133Cs固態NMR分析已用于評價Cs+在黏土礦物中的吸附位點和遷移[50-51]。Kim等[52]采用NMR譜研究了不同條件下Cs+在高嶺石和伊利石中的行為,得出Cs+在黏土礦物上的吸附受環境濕度、溶液濃度和Cs+滯留時間的影響,并推斷Cs+吸附的2個位點:基面位點和楔形位點。因此,NMR法有助于揭示放射性Cs+在黏土礦物中的吸附位點特性及吸附作用機制。

圖4 放射性Cs、Th/Eu調控金云母風化的機理Fig.4 Weathering of phlogopite controlled by Cs and Th/Eu
4.2 透射式電子顯微鏡
透射式電子顯微鏡(TEM)是研究Cs+與黏土礦物結合形態的有效工具,其原理是真空加速的電子束透過樣品,通過電磁場或靜電場收集到熒光板上,由于電子通過樣品傳輸,TEM能使樣品中的Cs原子成像。黏土礦物中的Cs+被視為黏土礦物結構的一部分。如,Mckinley等[53]觀測了Cs+在風化云母礦物上的FES;Fuller等[19]利用TEM拍攝了夾層和FES圖像并估算了夾層尺寸。研究發現,借助TEM分析可清晰地觀測FES與層間位點尺寸,但在FES和黏土礦物層間仍無法觀測到Cs+微粒或離子。此外,有學者利用TEM特別是高分辨透射電鏡(HRTEM)對黏土礦物的結構進行了分析,結果表明,層間與FES結構影響了Cs+的吸附[54-55]。Lee等[8]使用能譜透射電子顯微鏡(TEM-EDX)對Cs+和Ca2+在伊利石中的分布特征進行了研究,得出Cs+主要分布在邊緣位點,而Ca2+在伊利石顆粒中隨機分布的結論。雖然TEM能直觀反映黏土礦物結構,但其觀測范圍有限,需適當增加覆蓋整個黏土顆粒的TEM圖像范圍。
4.3 擴展X射線吸收精細結構光譜
近年來,擴展X射線吸收精細結構光譜(EXAFS)在環境領域廣泛應用,可從分子水平揭示放射性核素與黏土礦物的相互作用機制。EXAFS分析方法對研究Cs+與黏土礦物間的原子鍵長、原子間距、配位原子種類和數量等微觀結構信息作用明顯。Fan等[12]揭示了蛭石層間結構塌陷與Cs+吸附形態之間的關系,如圖5所示。Cs原子與氧原子發生配位反應,分別形成2個殼層,Cs+被水化第一殼層吸附,形成外層絡合(OSC)形態,Cs—O原子間距與Cs+在水溶液中的原子間距相當,鍵長2.95(蒙脫石)~0.32 nm(伊利石),配位數小于8,為弱吸附形態。相比之下,第2殼層為脫水吸附層,對于內層絡合(ISC),Cs—O/Si原子間距變大,鍵長為0.41~0.43 nm(0.45~0.47nm),配位數小于10,為強吸附形態。通過配位數之比可判定放射性Cs在不同黏土礦物上的賦存形態。

Cs—O1——Cs與土壤水中的氧發生配位;Cs—O2——Cs與四氧化硅中的氧發生配位圖5 EXAFS用于Cs+在蛭石礦物上吸附形態分析Fig.5 Speciation analysis of Cs+ adsorbed on vermiculite by EXAFS
Fan等[12]利用EXAFS技術與Tessier連續提取法,研究了Cs+與黏土礦物的作用機制。Cs+在伊利石中易形成ISC內層結合形態,具有強吸附作用而不易解吸;在外層的弱吸附位點形成OSC結合形態,弱吸附作用易解吸。作為中等膨脹性的蛭石黏土礦物,擁有大量的層間吸附位點,當Cs+進入層間結構后失去水合作用,導致蛭石層間晶格結構坍塌,最終形成穩定的晶格層間ISC,如圖6所示。有機質存在時,IS和ISC位點被隔離屏蔽,Cs+只能在黏土表面形成OSC。因此,土壤中存在的有機質不利于Cs+的吸附固定。
然而,EXAFS技術也有其局限性,通常需Cs+濃度大于50 mg/kg,自然界中放射性Cs含量極低,有的處于痕量水平,且黏土礦物的FES位點對Cs離子的吸附容量通常低于50 mg/kg,因此,EXAFS技術在低污染核素分析中有一定局限性。

圖6 土壤有機質與蛭石顆粒和Cs+的相互作用機制Fig.6 Interaction mechanism of soil organic matter on vermiculite particles and Cs+
4.4 放射自顯影
放射性核素顯像常用的3種方法分別是膠片自顯影、磷屏成像和使用微通道陣列探測器(MICAD)的電子放射自顯影(IP)。膠片自顯影是一種探測β粒子的方法,其原理是將膠片上的鹵化銀晶體顆粒中的銀原子還原成金屬銀,使膠片上顯示放射性同位素的自顯影圖像,再利用光學顯微鏡觀測放射性粒子[56]。IP技術是探測土壤中放射性粒子的有效方法。IP成像板網格經放大、立體纖維鏡掃描后,可實現顆粒物的定位和微觀結構分析,表征土壤微粒在成像板上的精確位置與輻射強度,成像板可重復利用,如圖7所示。該方法在土壤、植物組織、鳥類羽毛等樣品輻射或放射性Cs分布研究中得到了廣泛應用[57]。

圖7 土壤顆粒立體顯微鏡圖像Fig.7 Stereomicroscope image of soil particle
土壤中放射性Cs的濃度在10 ppb左右(10-10mol/L),該濃度已接近或低于常規核輻射儀器探測下限,而放射自顯影法可解決土壤中微量Cs離子濃度的測量技術瓶頸,通過測量單個黏土礦物顆粒中的輻射強度,評估土壤礦物對137Cs的吸附量。在不同土壤礦物類型、濃度、浸泡時間下,通過IP技術檢測到風化黑云母(WB)中137Cs的吸附容量明顯高于其他礦物,當137Cs濃度較低或反應時間較短時,僅從WB顆粒中檢測到137Cs,如圖8所示。實驗結果表明:WB在福島土壤中對放射性Cs有極強的吸附能力,證明了WB對放射性Cs的高富集特征[58]。將含有137Cs的WB顆粒添加至各種電解質溶液中,并通過IP技術可估算浸出前后放射性Cs的解吸量,隨著吸附時間的延長,137Cs離子沿WB層間區域遷移擴散,滯留在較為穩定的層間吸附位點,137Cs難以通過陽離子交換作用從層間結構解吸出來[59]。
為測定低放射性活度水平,膠片放射性自顯影通常需要很長的膠片曝光時間,膠片自顯影用于放射性定量的線性動態范圍僅1.5~3個數量級,很難確定樣品所需曝光時間,需要2次或多次曝光才能確定膠片線性動態范圍內的放射性活度,對低于該閾值的放射性沒有反應,且還存在一個飽和點,高于該點的β粒子和γ射線對膠片的光密度沒有額外作用,這些缺點限制了該技術的應用。
4.5 不同表征方法的特點與比較
NMR、TEM、EXAFS、IP等表征手段在土壤中放射性Cs吸附與解吸行為表征中均已得到應用,但各有其優缺點(表2),因此,在實際分析中應綜合多種表征技術,從宏觀與微觀2個層面揭示放射性Cs與黏土礦物的作用機制。

FB,新的黑云母;WB,風化黑云母;K,高嶺石;H,埃洛石;IL,伊利石;M,蒙脫石;A,水鋁英石;IM,伊毛縞石圖8 不同礦物顆粒吸附顯影成像Fig.8 Adsorption imaging of different mineral particles

表2 不同表征方法的優缺點Table 2 Advantage and disadvantage of different characterization methods
5 總結與展望
國外針對放射性Cs與黏土礦物的吸附、解吸及遷移等環境化學行為開展了深入研究。通過批式實驗開展吸附、解吸宏觀研究,針對不同土壤類型,特別是黏土礦物結構對吸附、解吸的作用機制不同,建立了多種吸附模型,其中等溫吸附模型、表面配位模型、多位點離子交換模型等得到不同程度的應用,多位點離子交換模型因其可用于解釋多數吸附、解吸過程,應用最為廣泛。但國內有關放射性Cs的環境行為研究較少,針對我國土壤類型的吸附模型的研究也鮮見報道。目前,放射性污染土壤為多種污染核素混合物,放射性Cs與土壤的吸附模型多為單個模型研究,單一的表征手段很難滿足吸附、解吸作用機制的研究需求。因此,建議可從以下幾個方面開展研究。
1) 開展土壤中放射性Cs與復雜體系污染核素(238U、90Sr、152Eu、239Pu等)共存時的相互調控作用研究,揭示相互調控機制。
2) 在放射性Cs與黏土礦物微觀表征分析技術方面,開展多種表征技術的綜合應用,特別是EXAFS、IP等先進表征技術,可從分子水平揭示放射性Cs與黏土礦物的微觀作用機制。
3) 現有的吸附模型多是在單一的黏土礦物結構吸附實驗基礎上提出的,現場土壤類型復雜,多數吸附模型難以滿足土壤吸附、解吸行為研究需求,應開展多模型融合研究,建立統一的土壤中放射性Cs吸附解吸行為模型。
4) 結構光譜與EXAFS分析是一種結構平均化的結果,在提取吸附產物中單個吸附構型的結構信息方面還存在一定的困難,可通過分子動力學以分子(原子或離子等)為研究對象[60],考察Cs離子在不同土壤結構中的微觀分子運動規律,解決實驗手段難以測定表面吸附結構的問題。
5) 針對實際放射性Cs污染土壤中Cs的吸附、解吸行為研究鮮見報道,應開展吸附、解吸模型在實際放射性Cs污染場地的驗證及應用研究。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
