液相色譜-三重四極桿/線性離子阱復合質譜技術檢測牛可食性組織中莫昔克丁的殘留
2021-03-01 06:18:58王亦琳王鶴佳
中國獸藥雜志 2021年2期
關鍵詞:方法
王亦琳,尹 暉,葉 妮,李 丹,孫 雷,王鶴佳
(中國獸醫藥品監察所,北京 100081)
莫昔克丁(moxidectin)又稱莫西菌素,是由一種鏈霉菌發酵產生的半合成單一成分的大環內酯類抗生素,是尼莫克丁(nemadectin)的乙酰胺化衍生物,屬于米爾貝霉素類抗寄生蟲藥物(Milbemycin)[1],其化學結構式見圖1。莫昔克丁具有較高的脂溶性,在體內主要分布于動物肝臟和脂肪組織中,且在體內殘留時間較長。莫昔克丁相對于伊維菌素和阿維菌素有更廣的驅蟲活性、長效性和安全性等特性[2]。20世紀80年代中期,莫昔克丁開始作為獸用驅蟲藥使用。作為新一代驅蟲藥物,莫昔克丁能夠高效的殺滅線蟲和體表寄生蟲,同時對動物有很好的安全性,對皮膚無刺激性。目前,莫昔克丁是被廣泛用于獸醫臨床的廣譜、高效、新型抗寄生蟲藥物。我國食品安全國家標準GB 31650-2019《食品中獸藥最大殘留限量》[3]規定,莫昔克丁在牛肌肉、肝臟、腎臟和脂肪組組織中的最高殘留限量(MRL)分別為20 μg/kg、100 μg/kg、50 μg/kg和500 μg/kg。
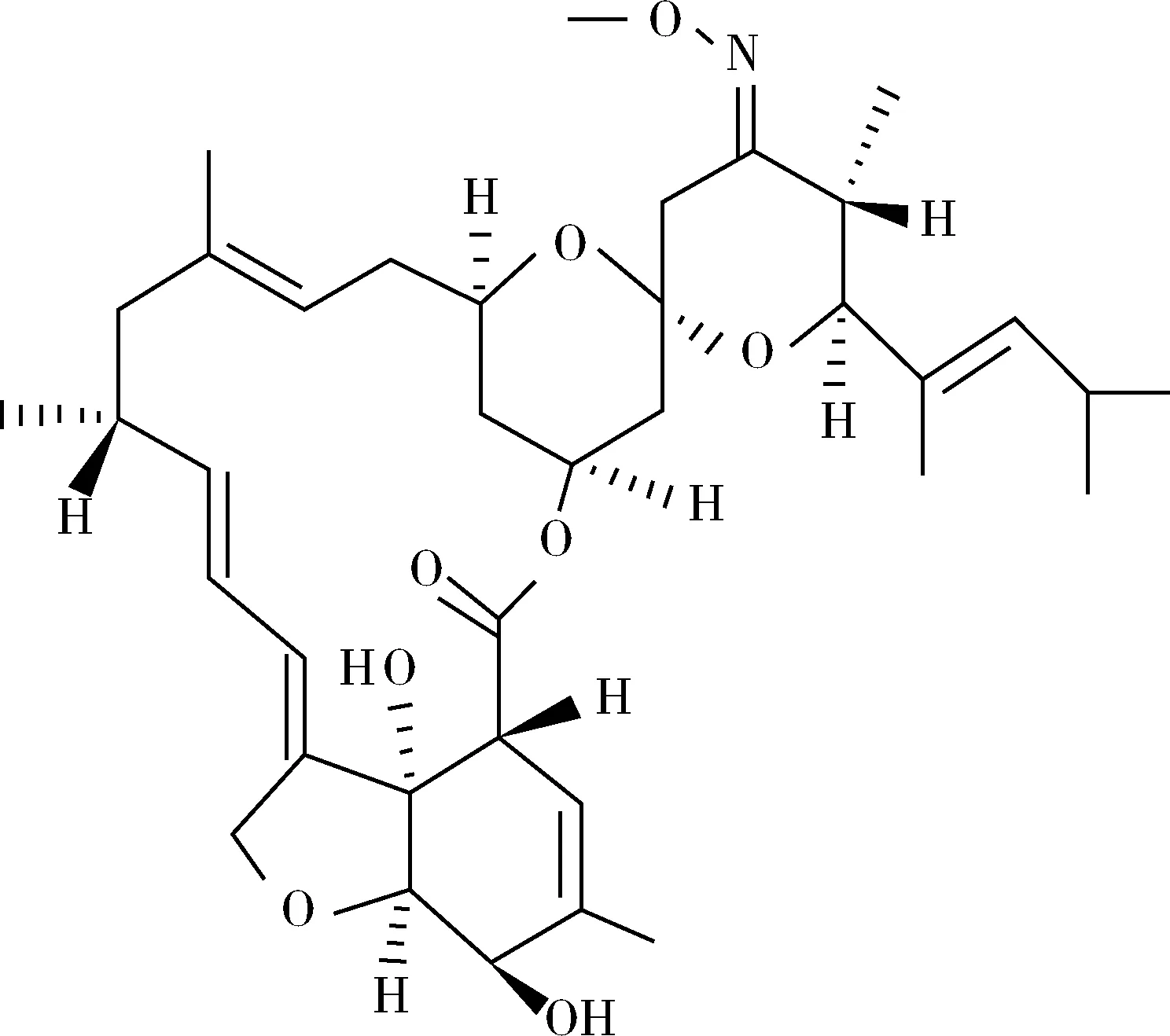
圖1 莫昔克丁化學結構式
目前國內外已經報道的動物性食品中莫昔克丁的殘留檢測分析方法多為液相色譜-熒光檢測法[4-6]。這種方法有較高的選擇性和靈敏度,但在樣品前處理過程中需要進行衍生化反應,步驟較為繁瑣,耗時較長。液相色譜-串聯質譜法的樣品前處理步驟相對于液相色譜來說一般較為簡單快速,不需要衍生化的過程[7-9]。液相色譜-三重四極桿/線性離子阱復合質譜技術相對于傳統的三重四極桿技術具有多反應監測聯合信息依賴性采集與增強子離子掃描(MRM-IDA-EPI)檢測模式,能夠對未知化合物進行雙重定性,大大排除了假陽性結果,提高了定性分析的準確度。本文建立了一種液相色譜-三重四極桿/線性離子阱復合質譜檢測方法,以滿足莫昔克丁在牛肌肉、肝臟、腎臟和脂肪等4種牛可食性組織中的殘留檢測要求。
1 材料與方法
1.1 儀器 日本島津公司Shimadzu 30A液相色譜儀-美國AB SCIEX公司Qtrap 6500質譜儀(配電噴霧離子源);AE260電子天平,Mettler Toledo公司;Biofuge Strators高速冷凍離心機,賀利氏公司;SIR4渦旋混合器,IKA公司;固相萃取裝置,Waters公司。
1.2 藥品和試劑 莫昔克丁對照品,購自德國Dr.Ehrenstorfer公司,含量≥94.0%;甲酸(色譜純)、甲醇(色譜純)、乙腈(色譜純),美國Fisher公司;C18固相萃取柱:300 mg/3 mL,美國Agilent公司;所用水為超純水。
1.3 標準溶液配制 精密稱取莫昔克丁對照品約10 mg,置于10 mL棕色量瓶中,用乙腈溶解并稀釋成濃度為1 mg/mL的莫昔克丁標準儲備液;精密量取1 mg/mL的莫昔克丁標準儲備液100 μL于10 mL量瓶中,用乙腈溶解并稀釋成濃度為10 μg/mL的莫昔克丁標準工作液;精密量取10 μg/mL的莫昔克丁標準工作液1 mL于10 mL量瓶中,用50%乙腈水溶液溶解并稀釋成濃度為1 μg/mL的莫昔克丁標準工作液。
1.4 樣品前處理
1.4.1 樣品的提取 稱取試料(2±0.02)g于50 mL離心管中,加乙腈6 mL,加入陶瓷均質子,渦旋30 s混勻后,中速水平振蕩5 min,10000 r/min離心5 min,轉移上清液至另一50 mL離心管中,加水10 mL,混勻,備用。
1.4.2 樣品的凈化與濃縮 C18固相萃取柱依次用乙腈5 mL和30%乙腈水溶液5 mL活化,取備用液8 mL過柱,用30%乙腈水溶液5 mL淋洗,抽干,用乙腈5 mL洗脫,收集洗脫液于10 mL試管中,于50 ℃水浴氮氣吹干。用50%乙腈水溶液1.0 mL充分溶解殘余物后過0.22 μm微孔濾膜,供液相色譜-三重四極桿/線性離子阱復合質譜測定。
1.5 儀器條件
1.5.1 色譜條件 色譜柱為BEH C18(50 mm×2.1 mm,粒徑1.7 μm),流動相A相為0.1%甲酸水溶液;B相為0.1%甲酸乙腈溶液,梯度洗脫:0~1 min保持75% B;1~3 min,75% B線性變化到95% B;3~4.5 min保持95% B;4.6~6 min保持75% B;流速:0.4 mL/min;柱溫:30 ℃;進樣量:5 μL。
1.5.2 質譜條件 電噴霧離子源(ESI+);電噴霧電壓為5500 V;離子源溫度為550 ℃;輔助氣1為55 psi;輔助氣2為55 psi;氣簾氣為30 psi;碰撞氣為High;檢測方式為多反應監測聯合信息依賴性采集與增強子離子掃描模式(MRM-IDA-EPI)。莫昔克丁定性、定量離子對及對應的去簇電壓、碰撞能量見表1。IDA參數:①使用動態背景扣除功能;②從不排除之前的目標離子;③觸發閾值:100 cps。EPI參數:①掃描范圍:m/z100~650;②掃描速度:10000 Da/s;③CE:35 ev;CES:15 ev。

表1 莫昔克丁MRM參數
1.6 基質匹配標準曲線的制備 精密量取適宜濃度的莫昔克丁標準工作液適量,用50%乙腈水溶液稀釋成濃度為1、2、5、10、20、50和100 ng/mL的系列標準工作液(適用于牛肌肉和腎臟);或1、2、5、10、20、50、100和200 ng/mL的系列標準工作液(適用于牛肝臟);或1、2、5、20、100、200、500和1000 ng/mL的系列標準工作液(適用于牛脂肪),從中各取1.0 mL,分別加入到空白試料經提取、凈化和吹干后的殘余物中,充分溶解,過微孔濾膜后作為基質匹配標準溶液上機測定。以特征離子質量色譜峰面積為縱坐標,基質匹配標準溶液濃度為橫坐標,繪制標準曲線,并計算回歸方程及相關系數。
1.7 方法準確度及精密度的測定 采用標準添加法,在空白牛肌肉、肝臟、腎臟和脂肪中添加定量限、MRL、2MRL三個濃度的莫昔克丁標準工作液。每種濃度5個樣品平行試驗,重復3次,按照1.4樣品前處理方法處理之后上機測定,基質匹配標準溶液外標法定量,計算回收率、批內、批間變異系數。
1.8 方法靈敏度確定 添加適量濃度的莫昔克丁標準工作液于2 g空白牛肌肉、肝臟、腎臟和脂肪組織中,經前處理后測定,觀察藥物特征離子質量色譜峰信噪比(S/N)和對應藥物濃度,以 S/N>10(按PtP算)作為方法的定量限。
2 結果與分析
2.1 定性分析 在日常的MRM檢測模式中,通常選擇一個母離子和兩個響應較強的子離子進行定性分析。但在殘留檢測的實際工作中,由于待測樣品通常為各種動物組織樣品,基質比較復雜,盡管通過樣品前處理進行了凈化操作,檢測中仍會出現基質干擾峰,影響定性和定量的準確性。本研究采用MRM-IDA-EPI檢測模式,可以在利用MRM精確定量的同時,得到目標子離子的二級子離子全掃描質譜圖(EPI圖),通過與對照溶液的EPI圖進行對比,可以更為準確地做出定性判斷。圖2是莫昔克丁陽性添加樣品和對照溶液的EPI圖對比,可以發現兩者的特征子離子匹配度較高,呈現出子離子“指紋圖”的效果。按照一個母離子1個識別點(IP)點,一個子離子1.5個IP點計算,圖2中主要有7個特征子離子,共11.5個IP點,超出歐盟2002/657/EC[10]規定的至少3個IP點的要求,進一步提高了生物樣本復雜基質中定性的準確性。
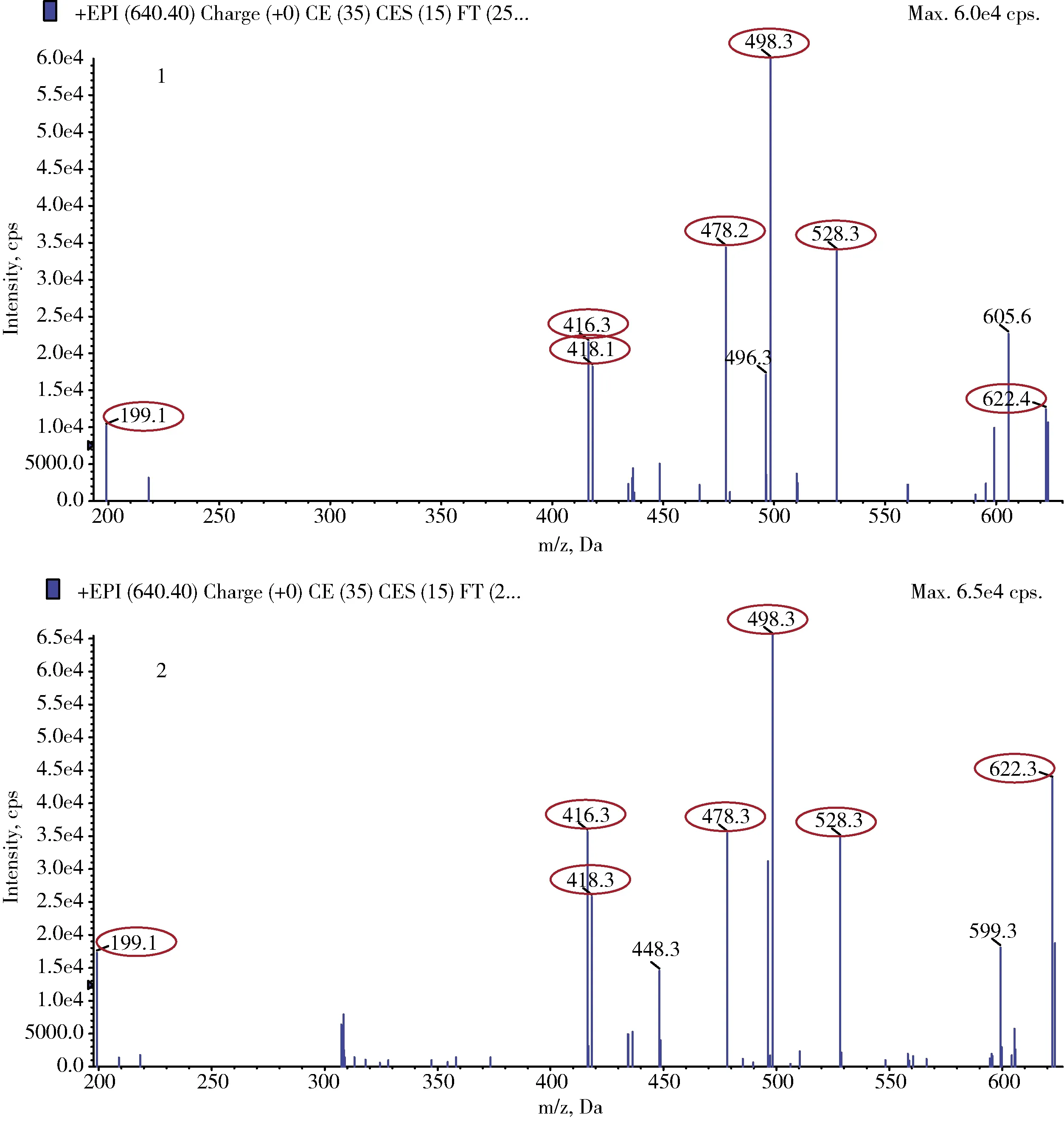
1.莫昔克丁空白牛肝添加樣品;2.莫昔克丁對照溶液
2.2 基質匹配標準曲線 以莫昔克丁的特征離子質量色譜峰面積與其對應的基質匹配標準溶液濃度作圖,得到相應的標準曲線,線性回歸方程及相關系數(R2)見表2。莫昔克丁在牛肌肉和牛腎臟1~100 ng/mL的基質匹配標準溶液濃度范圍內;在牛肝臟1~200 ng/mL的濃度范圍內和在牛脂肪1~1000 ng/mL的基質匹配標準溶液濃度范圍內,特征離子質量色譜峰面積與濃度均呈良好的線性關系,R2均>0.990。

表2 莫昔克丁基質匹配標準曲線的回歸方程和相關系數
2.3 方法靈敏度 按1.8中所述方法進行處理,依據特征離子質量色譜峰信噪比S/N>10為方法的定量限,得出在空白牛肌肉、肝臟、腎臟和脂肪組織中,莫昔克丁的定量限均為2 μg/kg。2 μg/kg空白牛肌肉、肝臟、腎臟和脂肪組織添加樣品中莫昔克丁的特征離子質量色譜圖見圖3。
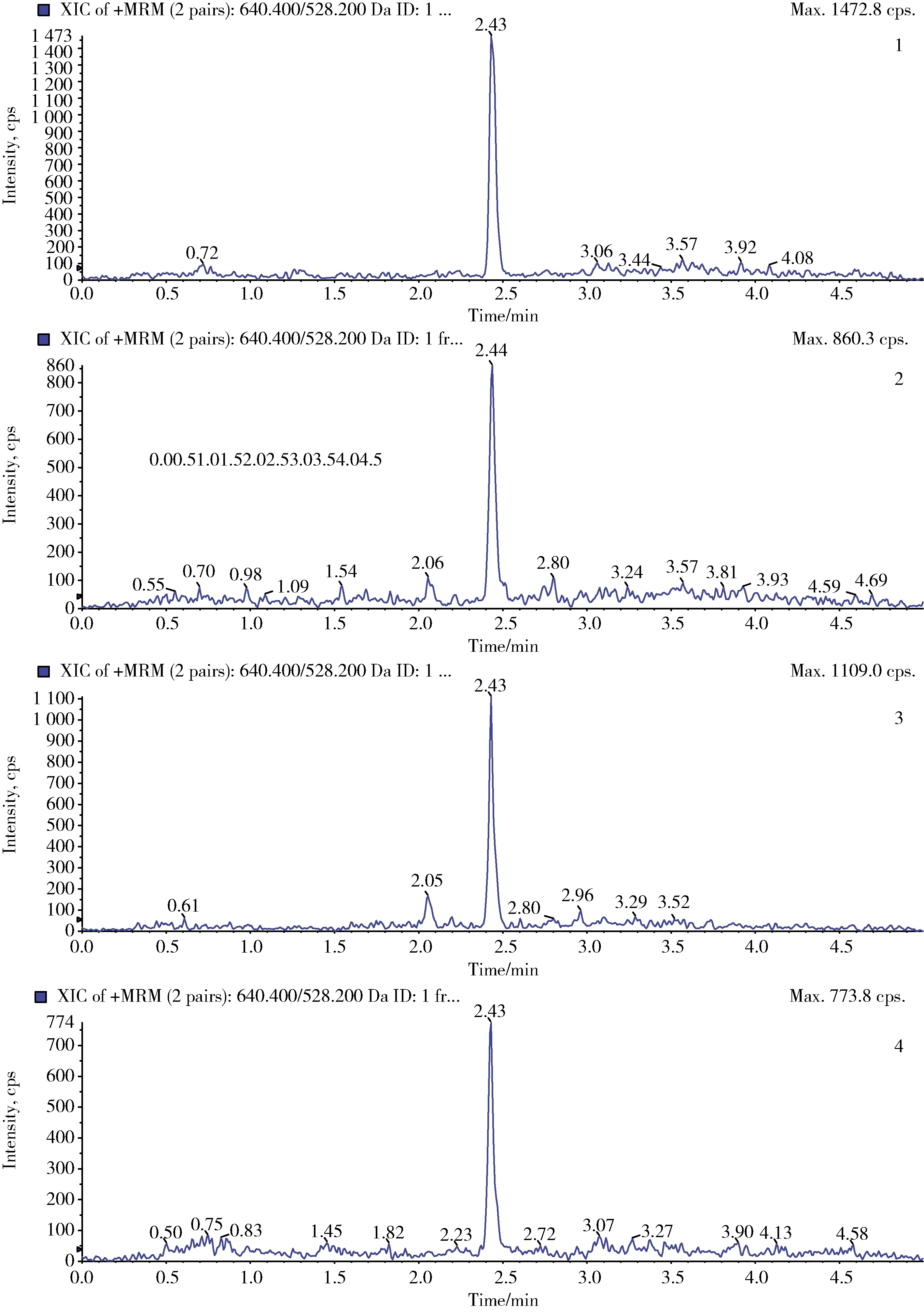
1.牛肌肉;2.牛肝臟;3.牛腎臟;4.牛脂肪
2.4 方法準確度及精密度 在空白牛肌肉、肝臟、腎臟和脂肪中各添加3個不同濃度的莫昔克丁標準工作液進行回收率試驗,結果匯總見表3,莫昔克丁在牛肌肉2~40 μg/kg、牛肝臟2~200 μg/kg、牛腎臟2~100 μg/kg和牛脂肪2~1000 μg/kg添加濃度水平上的回收率在65.6%~115%范圍內;批內與批間RSD均小于15%。結果表明本方法定量準確,重復性好。
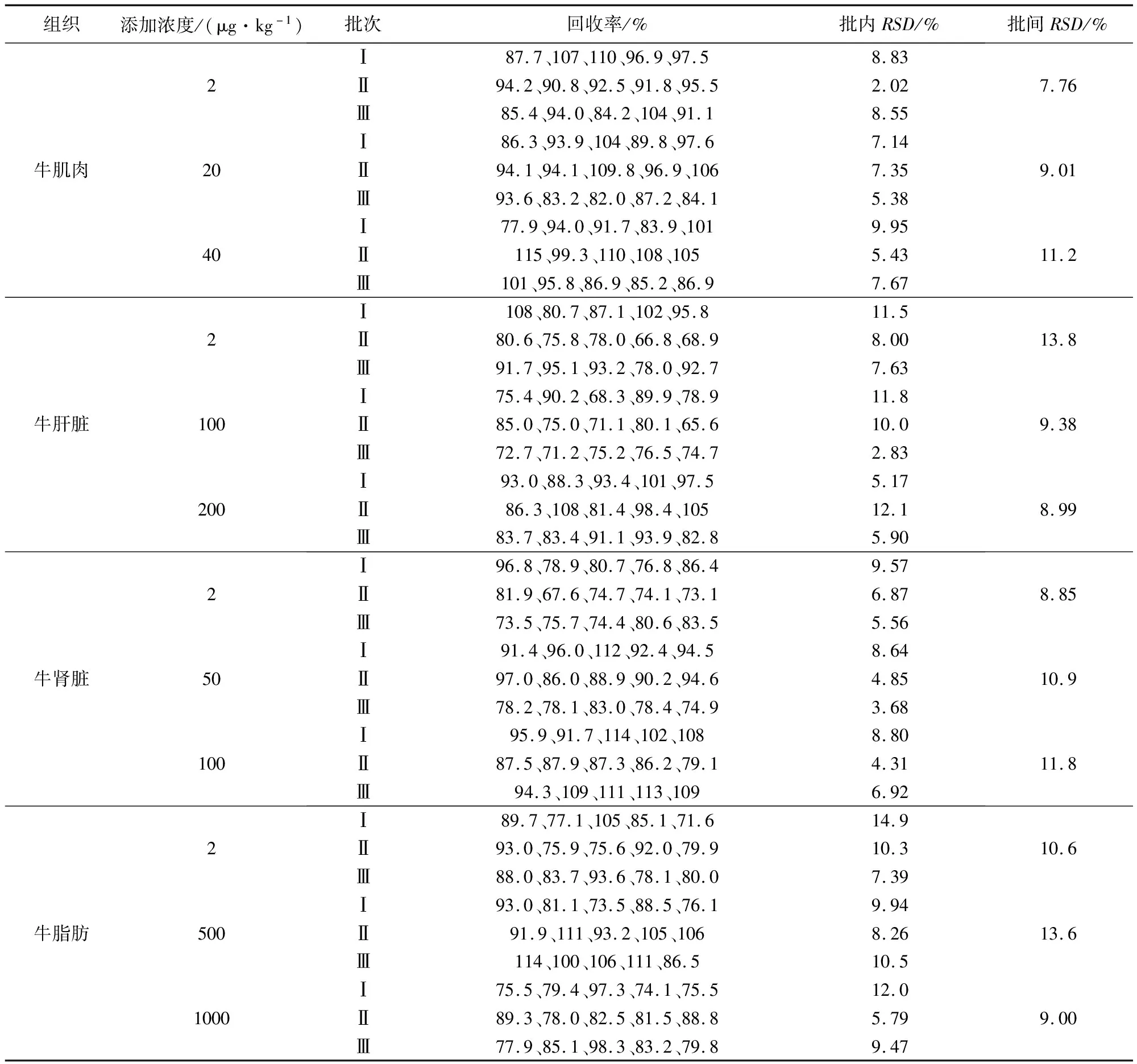
表3 空白牛可食性組織中莫昔克丁添加的回收率(n=5)
3 討論與結論
3.1 樣品前處理方法的建立 通過大量查閱國內外文獻,試驗中最先參考已報道的幾個QuEChERS前處理方法[11-12]:使用乙腈提取,MgSO4除水,PSA粉末凈化。結果發現這種方法處理牛肉回收率較好,但處理牛肝回收率較低,且譜圖中有很多干擾的雜峰,說明該方法對牛肝這種基質更為復雜的樣品凈化效果不佳。考慮到莫昔克丁分子結構的母核與阿維菌素和多拉菌素有相似之處[13],又參考了阿維菌素和多拉菌素的前處理方法[14-15]:乙腈提取后加水稀釋,用三乙胺調節pH值,經C18固相萃取柱凈化。但是在實驗中發現,當提取液中加入三乙胺時,會明顯降低方法的回收率,因此不使用三乙胺。同時我們也考察了C18柱對莫昔克丁的保留效果和不同濃度的乙腈水溶液的淋洗效果,最終確定了用30%乙腈水溶液淋洗,可以在有效去除樣品中極性雜質的同時,不會將莫昔克丁藥物洗脫造成回收率損失。考慮到過柱的時間因素,將提取溶液的一半進行過柱,能夠較為有效的去除雜質,同時減少一半的基質影響,最后定容體積也采用了1.0 mL的體積,而不是濃縮的0.5 mL體積,降低了氮氣吹干后濃縮步驟的影響。
3.2 質譜條件的建立和定性方法的選擇 由于本文的分析對象牛肌肉、肝臟、腎臟、脂肪均屬于動物源性樣品,基質比較復雜,在使用一般的MRM方法進行樣品檢測時,目標分析物容易受到復雜基質的干擾影響。樣品中定性離子對豐度比與對照溶液相比,容許偏差易超出歐盟2002/657/EC[10]中的規定,影響定性的準確性。因此我們建立了MRM-IDA-EPI的質譜分析方法。首先使用針泵進樣,在正離子模式下進行Q1掃描,找到準分子離子峰。之后對準分子離子峰進行子離子掃描,選擇相應值最強且穩定的兩個子離子分別作為定量離子和定性離子,并進一步優化MRM各項參數。接著我們利用儀器的線性離子阱(Trap)功能在第三重四極桿處(Q3)進行二級子離子的富集掃描[16-17]。使用動態背景扣除功能,并將閾值設為100 cps。在掃描過程中當MRM通道采集的信號超過100 cps時就會觸發EPI增強子離子掃描。CE和CES分別設為35 ev和15 ev,可以同時得到20、35和50 ev三個碰撞能量水平下的子離子質譜圖,獲得更為全面的質譜信息。該檢測方法在使用MRM定量的同時,可以得到目標子離子的二級質譜全掃描圖。在實際樣品檢測時,可將樣品與基質匹配標準溶液的二級子離子圖進行對比,比較兩者的特征離子的分布及強度,進而做出更為準確地判斷。該方法豐富了樣品定性所需要的質譜數據,提高了方法的選擇性[18]。
3.3 定量方法的選擇 該方法針對牛肌肉、肝臟、腎臟、脂肪,在進行前處理后上機測試發現均存在一定的基質效應。因此在定量方法選擇時采用了基質匹配標準溶液法進行定量,可以有效消除基質效應的影響,提高方法定量的準確性。
本文采用液相色譜-三重四極桿/線性離子阱復合質譜技術建立了一種可檢測牛肌肉、肝臟、腎臟和脂肪中莫昔克丁殘留的分析方法。該方法在進行常規的MRM采集同時,采用IDA-EPI模式又可以獲得目標分析物子離子的完整二級質譜信息,同時實現了定性和定量,可以有效防止假陽性的產生。同時該方法具有良好的可操作性和重現性,方法靈敏度、準確度和精密度均能滿足獸藥殘留分析方面的要求。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
