脊髓性肌萎縮癥SMN1基因2+0基因型攜帶者的家系研究
2021-02-26 00:00:48曹延延程苗苗宋昉瞿宇晉白晉麗金煜煒王紅
遺傳 2021年2期
曹延延,程苗苗,宋昉,瞿宇晉,白晉麗,金煜煒,王紅
研究報告
脊髓性肌萎縮癥基因2+0基因型攜帶者的家系研究
曹延延,程苗苗,宋昉,瞿宇晉,白晉麗,金煜煒,王紅
首都兒科研究所遺傳室,北京 100020
脊髓性肌萎縮癥(spinal muscular atrophy, SMA)是一種兒童時期較為常見的神經肌肉病,屬于常染色體隱性遺傳。絕大多數SMA由運動神經元存活基因1 (survival motor neuron 1,)的純合缺失突變所致。而的2+0基因型個體作為一種特殊的SMA攜帶者,給攜帶者篩查以及家系的遺傳咨詢帶來了巨大的挑戰。已有研究表明,g.27134T>G和g.27706_27707delAT多態位點變異對于Ashkenazi猶太人群中的2+0基因型個體具有提示作用。為進一步探究這兩個多態位點是否在中國人群也具有特異性,本研究納入了44例家系成員和204例已知基因拷貝數的對照樣本。44例家系成員來自于9個無關的基因純合缺失的SMA家系,先證者雙親之一疑似為2+0基因型攜帶者。利用多重連接探針擴增(multiplex ligation-dependent probe amplification, MLPA)和短串聯重復(short tandem repeat, STR)連鎖分析進行基因型的鑒定以及多態位點的篩查,最終通過對家系三代成員或多子女家系兩代成員的分析確定了9個家系中的10例個體為2+0基因型攜帶者,多態位點篩查顯示1例攜帶3拷貝基因的個體同時存在g.27134T>G和g.27706_27707delAT多態位點的變異。因此,本研究通過對2+0基因型攜帶者的鑒定,為家系遺傳病的診斷提供了精準的遺傳咨詢。g.27134T>G和g.27706_27707delAT多態位點可能與中國人群2+0基因型個體的關聯度較低,尚需尋找中國人群特異的多態位點以提高2+0基因型攜帶者的檢出率。
脊髓性肌萎縮;基因;2+0基因型;STR連鎖分析;多態位點
脊髓性肌萎縮癥(spinal muscular atrophy, SMA)是兒童時期較為常見的神經肌肉病,呈常染色體隱性遺傳。運動神經元存活基因1 (survival motor neuron 1,)是SMA的致病基因,絕大部分的SMA患兒是由于基因的純合缺失突變所致。基因與基因高度同源,是SMA的表型修飾基因。此外,基因和基因均定位于5q13.2,基因靠近端粒,而基因靠近著絲粒,二者成鏡像排列。這些特征使得基因易于發生缺失、重復以及與之間的基因轉換,造成人群中基因拷貝數變異較大[1]。
正常情況下,每條染色體攜帶1拷貝基因和1拷貝基因,即每個個體的基因和基因的拷貝數均為2。對于基因純合缺失的先證者(拷貝數為0),其雙親基因的拷貝數通常為1,也就是一條染色體上的基因丟失,即經典的SMA攜帶者。但是SMA還有一種特殊類型的攜帶者,這類攜帶者的基因拷貝數雖然為2,但是卻位于同一條染色體(in),而另一條染色體上的基因丟失,即2+0型攜帶者。有效識別2+0型攜帶者對于提高SMA攜帶者檢出率以及為家庭提供精準的遺傳咨詢至關重要。
已有研究顯示在Ashkenazi猶太人群[2],大部分攜帶有重復等位基因的個體(拷貝數大于等于3)可存在一種由g.27134T>G和g.27706_ 27707delAT多態位點構成的單倍型。由此推測在特定人群中,這兩個多態變異的存在對于2+0基因型攜帶者具有提示作用。
本研究納入9個無關的中國SMA家系,先證者雙親之一疑似2+0型攜帶者。通過基因(和基因)劑量分析結合短串聯重復(short tan-dem repeat, STR)連鎖分析明確疑似2+0攜帶者的基因型,并初步探討g.27134T>G和g.27706-27707delAT多態位點是否適于中國人群2+0基因型的預測。
1 對象與方法
1.1 研究對象
本研究納入了9個基因純合缺失的SMA家系,共44例家系成員。其中,SMA先證者9例,先證者同胞2例,先證者雙親18例,先證者祖父母/外祖父母15例。所有先證者均為在首都兒科研究所遺傳室加入“中國罕見病注冊登記研究—SMA注冊登記”項目(編號:2016YFC0901505)的患者,且所有參與家庭均簽署知情同意書。本研究經首都兒科研究所倫理委員會批準(批準號:SHERLL2017007)。此外,另加入204例已知基因拷貝數的本研究室貯存DNA對照樣本用于多態位點的篩查。
1.2 拷貝數分析
先證者及家系成員留取EDTA抗凝血3 mL,采用?Blood Genomic DNA Kit (北京全式金生物技術有限公司)提取基因組DNA。應用多重連接探針擴增(multiplex ligation-dependent probe ampli-fication, MLPA)技術(MRC,荷蘭)進行基因拷貝數和多態位點的分析。其中,P060-B2 SMA Kit用于測定基因和基因的拷貝數,P460-A1 SMA Kit用于多態位點g.27134T>G和g.27706-27707delAT的檢測。
1.3 STR連鎖分析
選取12個STR位點應用毛細管電泳的方法進行連鎖分析,其中6個位點(UHM2、UHM3、UHM4、UHM5、UHM7和UHM8)位于基因上游2 Mb范圍內,其余6個位點(DHM1、DHM2、DHM4、DHM6、DHM7和DHM8)位于基因下游2Mb范圍內。家系的STR連鎖分析實驗及數據處理由上海五色石醫學研究股份有限公司完成。
2 結果與分析
2.1 SMN1基因拷貝數分布
9例SMA先證者的基因拷貝數均為0,即為基因純合缺失。先證者的雙親之一均攜帶2拷貝基因(3例父親;6例母親),其配偶基因的拷貝數均為1,即為基因雜合缺失。先證者的15例祖父母或外祖父母中,8例個體攜帶3拷貝基因,除1例僅有單方樣本外,其余7例的配偶均為基因雜合缺失。最后,在2例SMA先證者的同胞中基因拷貝數分別為2和3 (表1)。
2.2 SMN1基因型的分布
通常情況下,正常個體每條染色體攜帶1拷貝基因,即1+1基因型。0+0基因型表示兩條染色體上的基因均丟失,即純合缺失突變,為SMA患者。1+0基因型表示一條染色體上基因拷貝數為1,另一條染色體上的基因丟失,即經典的SMA攜帶者;2+0基因型表示2拷貝的基因位于同一條染色體,另一條染色體上基因丟失,即為特殊類型的SMA攜帶者。2+1基因型表示一條染色體攜帶2拷貝的基因,而另一條染色體上攜帶1拷貝的基因,基因的總拷貝數為3。2+0基因型和2+1基因型個體為重復等位基因的攜帶者,二者表型正常。
進一步分析基因的基因型分布(表1),9例先證者均為0+0基因型,即基因純合缺失。先證者的雙親中,50%(9/18)為經典型SMA攜帶者(1+0基因型),同時另一方(50%,9/18)均為疑似2+0型的攜帶者。同樣,在3對祖父母和4對外祖父母中,一方(50%, 7/14)為經典1+0攜帶者,另一方(50%, 7/14)為2+1基因型。另有1例外祖父(僅有單方樣本)為2+1基因型。2例先證者同胞中,基因型分別為2+0和2+1。
2.3 SMN1基因拷貝數的家系分布
9個家系均進行了包括先證者、同胞、父母、祖父母或外祖父母在內的和基因拷貝數檢測以及STR連鎖分析,以明確先證者攜帶2拷貝基因雙親的基因型(表2)。
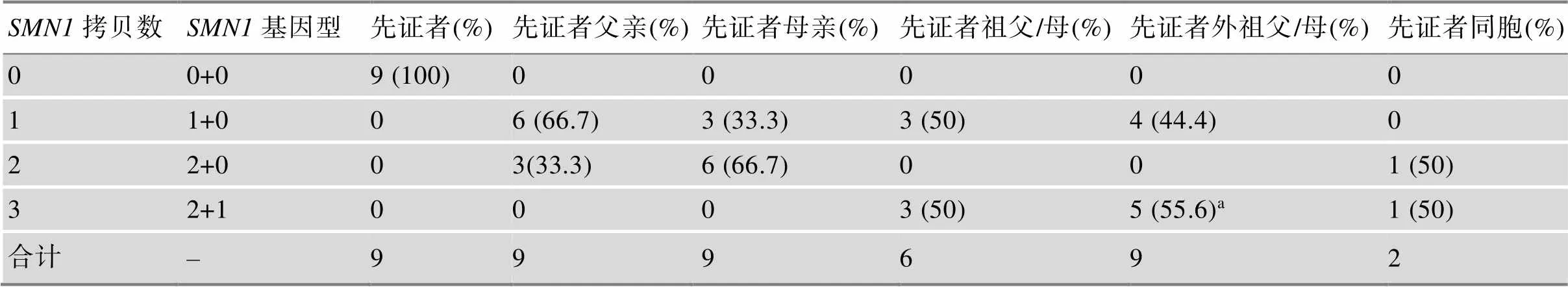
表1 SMN1拷貝數和基因型的分布
a:僅單方樣本,其配偶離世。
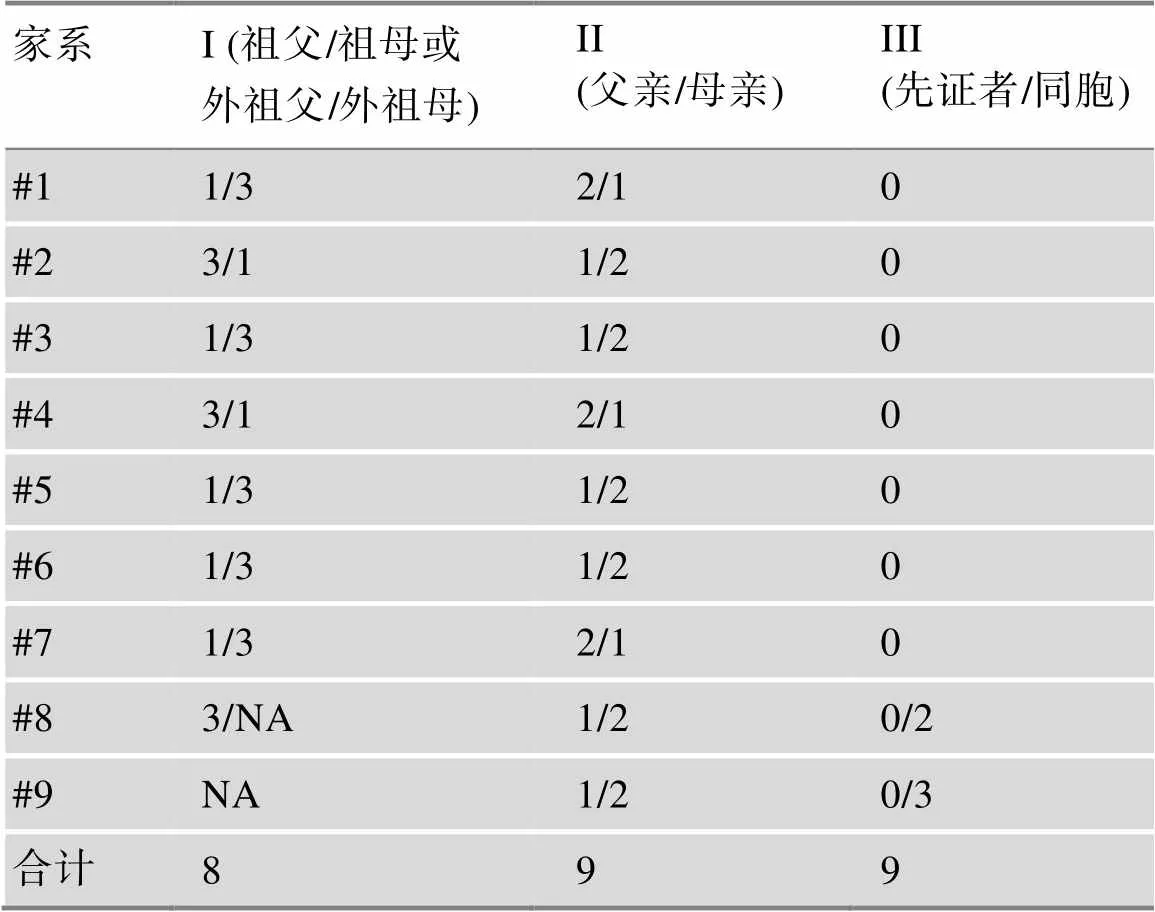
表2 SMN1基因拷貝數在家系中的分布
NA:無法獲得樣本;I,II和III:代系。
以#7號家系為例,先證者基因純合缺失,母親為基因的雜合缺失,父親基因的拷貝數為2 (圖1)。因此,先證者父親疑似為2+0基因型。此外,先證者的祖父和祖母基因的拷貝數分別為1和3。結合STR連鎖分析顯示,父親攜帶黑色0拷貝的基因遺傳自祖父,攜帶綠色2拷貝的基因遺傳自祖母。隨后父親將黑色0拷貝的基因傳遞給先證者,同時母親也將橙色0拷貝的基因傳遞給先證者,導致先證者發生SMA。因此,該家系通過三代基因拷貝數結合STR連鎖分析,基本確定了先證者父親基因為2+0型。同時也基本排除了先證者基因的新生變異。
此外,值得注意的是#8和#9家系。#8家系中先證者的外祖母由于過世沒有獲得樣本,該家系有兩個孩子。如圖2所示,先證者母親為2+0基因型,其藍色的2拷貝基因遺傳自先證者的外祖父,推測橙色的0拷貝基因可能遺傳自先證者的外祖母。隨后母親將橙色的0拷貝基因傳遞給先證者,加之另一個遺傳自先證者父親的黑色0拷貝基因,先證者發病。但是,先證者母親將藍色的2拷貝基因傳遞給了先證者的姐姐,而姐姐也同時繼承了父親黑色0拷貝基因。因此,姐姐的基因拷貝數雖然為2,但是她實際與母親一樣,為2+0攜帶者。
#9家系,該家系中先證者的外祖父母均已過世,家中有兩個孩子。如圖3所示,先證者為基因純合缺失,父親為基因雜合缺失,母親的基因拷貝數為2。因為無法得到先證者外祖父和外祖母的樣本,因而不能通過三代連鎖分析確定母親的基因型。母親基因為2拷貝主要有兩種可能:2+0基因型攜帶者或母親為正常的1+1基因型。鑒于先證者哥哥基因的拷貝數為3,STR連鎖分析顯示,哥哥的綠色1拷貝基因遺傳自父親,另2拷貝的基因只能來自母親,故而推測出另2拷貝的基因位于同一條染色體。因此,通過家系中的多子女連鎖分析也可確定先證者母親為2+0基因型。
2.4 多態位點分析
為明確多態位點g.27134T>G和g.27706_ 27707delAT是否可用于中國人群2+0基因型的預測,本研究篩查了44例家系成員以及204例攜帶不同基因拷貝數的對照樣本。
本研究中的44例家系成員,基因型為0+0的有9例,1+0基因型16例,經實驗確定的2+0基因型10例,還有2+1基因型9例。這些樣本中均未發現g.27134T>G和g.27706_27707delAT多態位點變異。考慮到這些家系成員間存在親緣關系,我們在204例無親緣關系且已知基因拷貝數的對照樣本中對這兩個多態位點進行了篩查。對照樣本中,187例攜帶2拷貝基因,其中包括18例疑似2+0基因型個體。這18例個體的子代均為基因純合缺失的SMA患兒,配偶均為基因雜合缺失的SMA攜帶者。此外,對照樣本中還包括16例攜帶3拷貝基因個體和1例攜帶4拷貝基因的個體。結果顯示,有1例基因拷貝數為3的個體同時存在g.27134T>G和g.27706_ 27707delAT多態位點變異,而其余樣本未發現攜帶有這兩個位點的多態性改變。(表3)。
3 討論
對于純合缺失突變的SMA家系來說,當雙親一方為經典1+0攜帶者,而另一方基因拷貝數為2時,明確雙親的基因型對于家庭SMA再發風險的評估至關重要。若攜帶2拷貝基因的雙親一方為2+0基因型,那么他們再次生育,子代SMA的再發風險為25%;若為正常的1+1基因型,那么先證者可能發生了新生變異抑或是雙親之一為生殖腺嵌合體,因而該家系SMA的再發風險很低。本研究通過對家系三代成員,或多子女家系的兩代成員進行基因拷貝數檢測,并結合基因上下游STR位點的家系連鎖分析最終確定了來自9個家系的10例個體為基因的2+0攜帶者。同時,也明確了這9個家系中的先證者非基因的新生變異所致,為該家系提供精準的遺傳咨詢奠定了基礎。
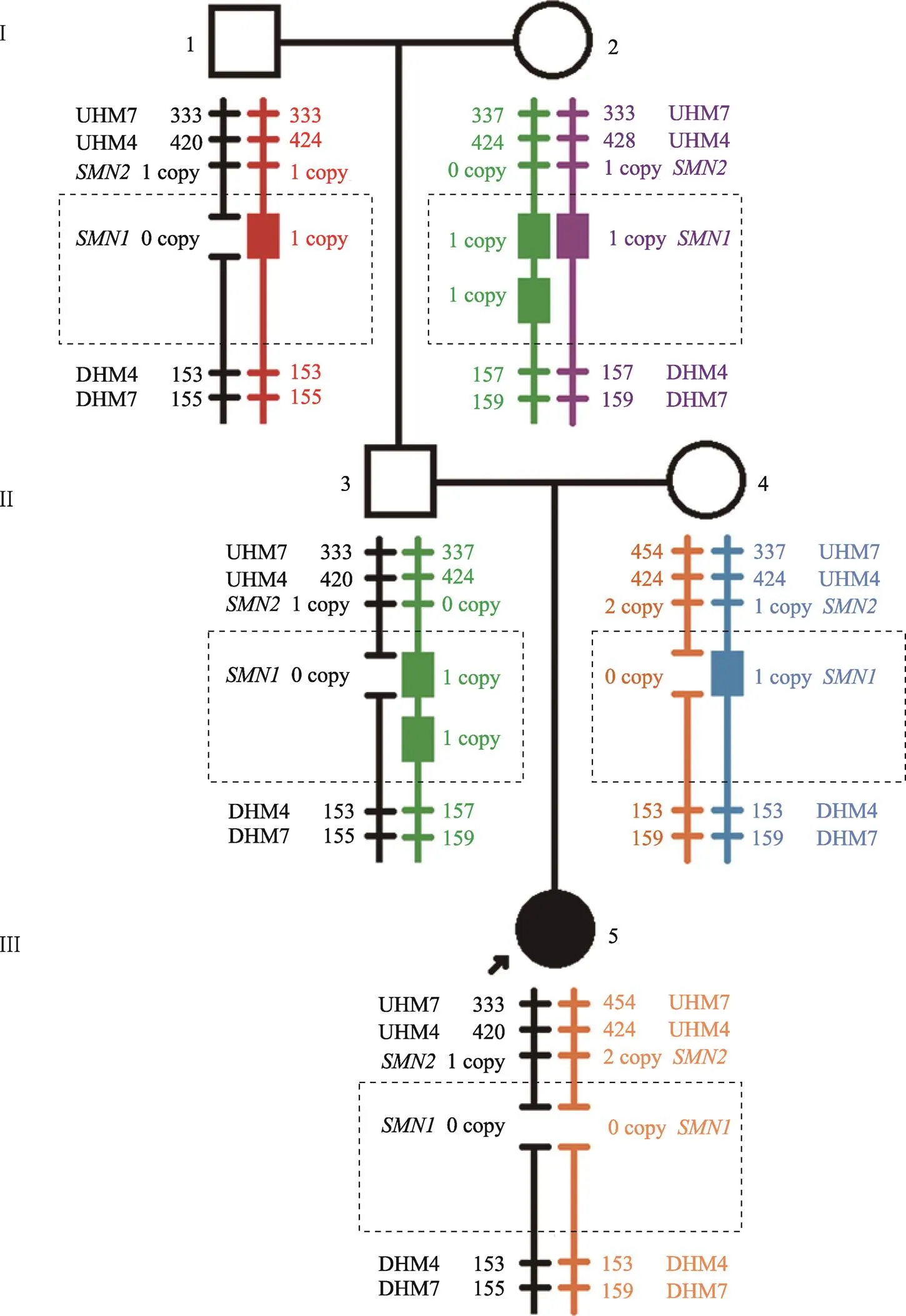
圖1 #7家系SMN基因拷貝數與STR連鎖分析模式圖
I:祖父/祖母;II:父親/母親;III:先證者;正方形:男性;圓形:女性;黑色圓形:女性先證者;不同顏色的豎線:不同來源的染色體;短橫線及數字:STR位點及其片段長度;實心長方形:1拷貝的基因;兩短橫線間空白:0拷貝的基因;虛線框:基因的總拷貝數;UHM7:上游單倍型標記7;UHM4:上游單倍型標記4;DHM4:下游單倍型標記4;DHM7:下游單倍型標記7。
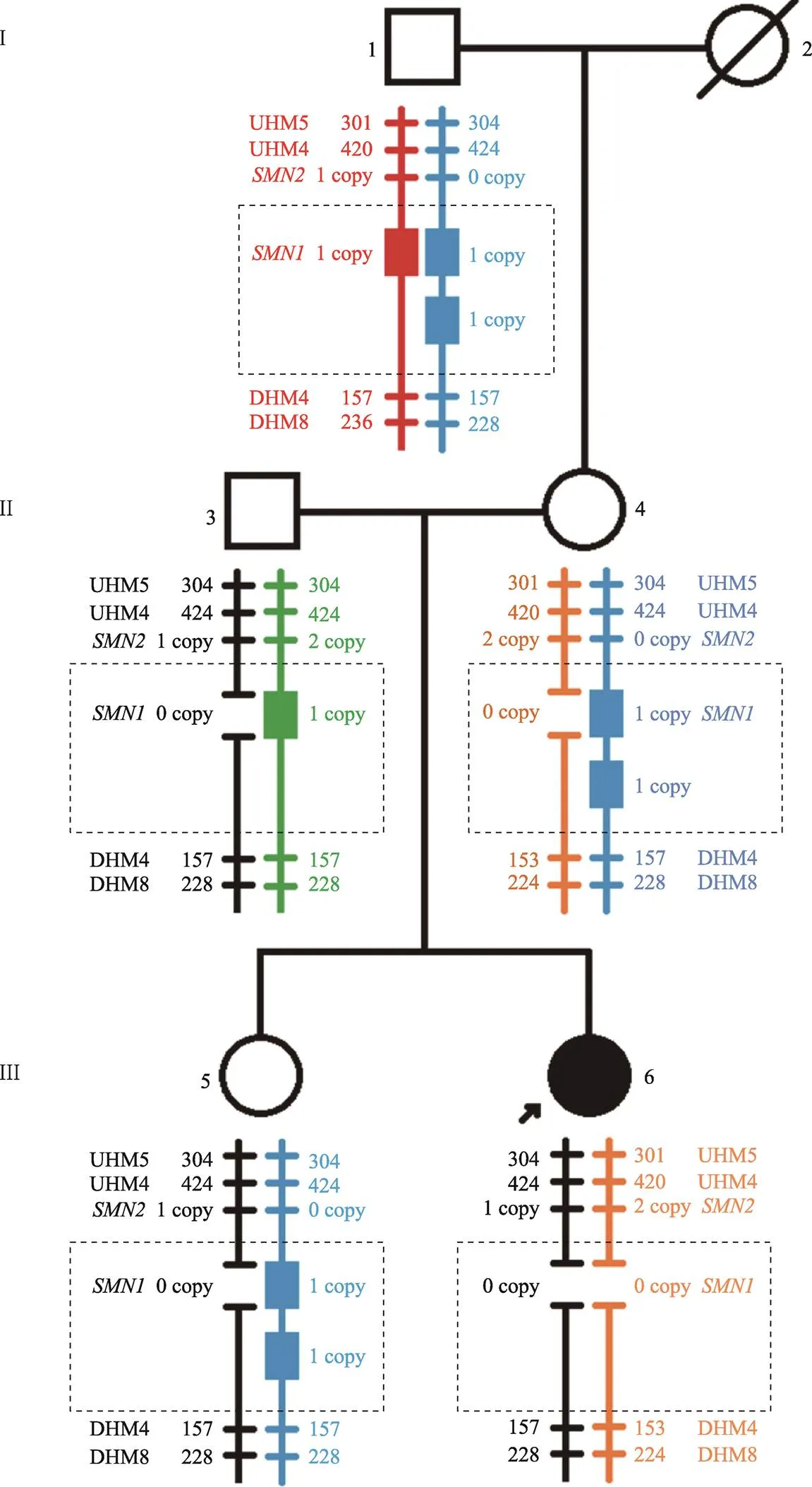
圖2 #8家系SMN基因拷貝數與STR連鎖分析模式圖
I:外祖父/外祖母(外祖母已去世);II:父親/母親;III:先證者/同胞;正方形:男性;圓形:女性;黑色圓形:女性先證者;不同顏色的豎線:不同來源的染色體;短橫線及數字:STR位點及其片段長度;實心長方形:1拷貝的基因;兩短橫線間空白:0拷貝的基因;虛線框:基因的總拷貝數;UHM5:上游單倍型標記5;UHM4:上游單倍型標記4;DHM4:下游單倍型標記4;DHM8:下游單倍型標記8。
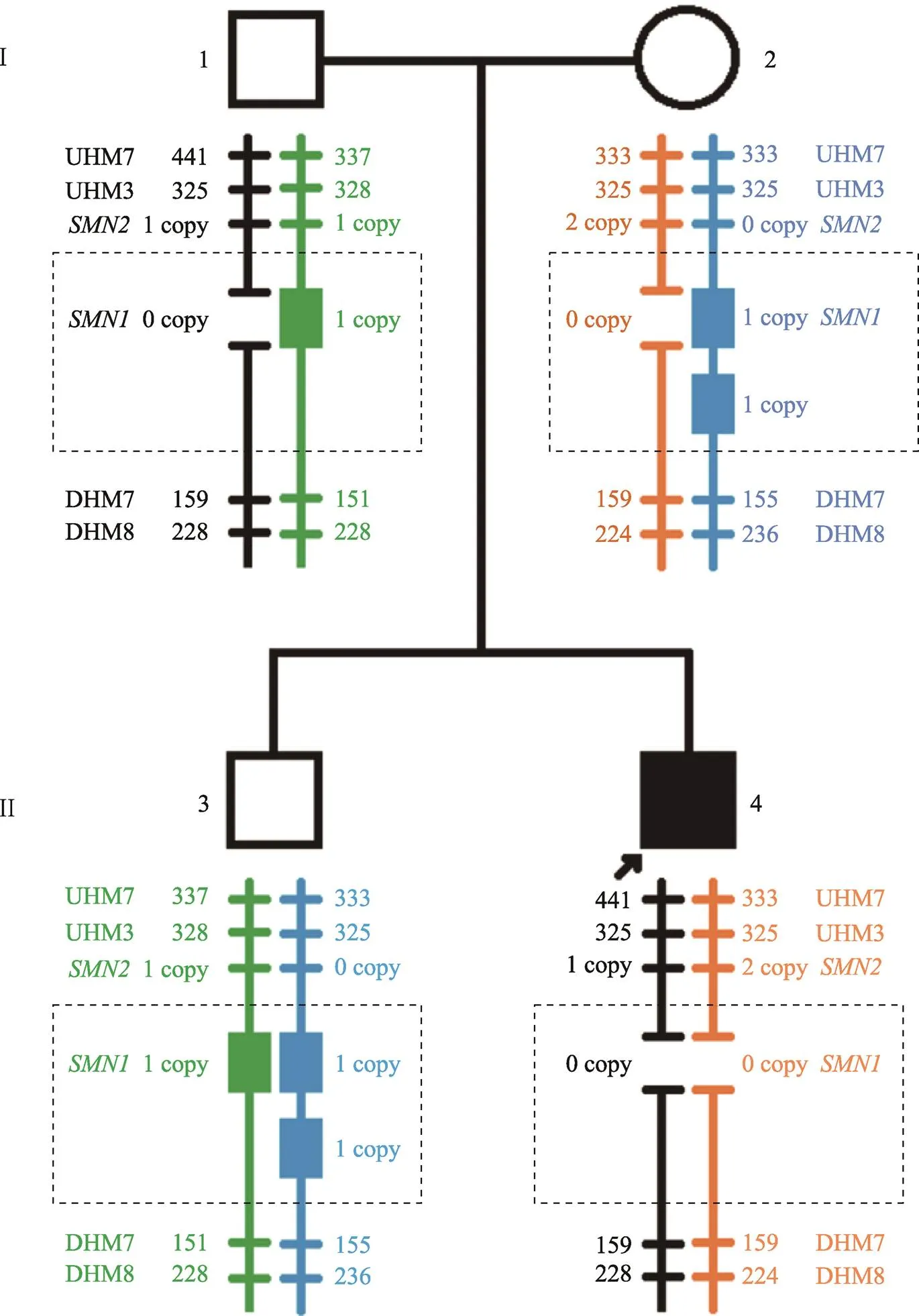
圖3 #9家系SMN基因拷貝數與STR連鎖分析模式圖
I:父親/母親;II:先證者/同胞;正方形:男性;黑色正方形:男性先證者;圓形:女性;不同顏色的豎線:不同來源的染色體;短橫線及數字:STR位點及其片段長度;實心長方形:1拷貝的基因;兩短橫線間空白:0拷貝的基因;虛線框:基因的總拷貝數;UHM7:上游單倍型標記7;UHM3:上游單倍型標記3;DHM7:下游單倍型標記7;DHM8:下游單倍型標記8。
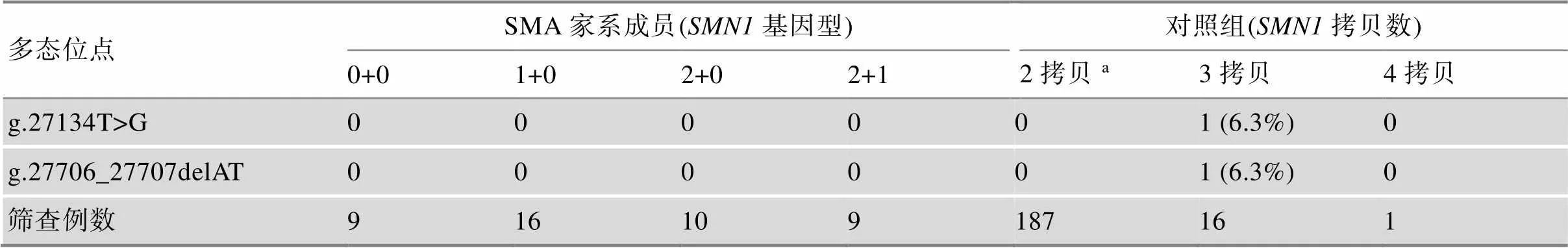
表3 多態位點的分布
a:包含18例疑似2+0基因型個體,即先證者為基因純合缺失,雙親之一為雜合缺失,另一方基因拷貝數為2(為疑似2+0基因型)。
作為特殊類型的攜帶者,2+0基因型在一般人群中的攜帶率約為5%~8%[3],在肯定攜帶者中約占4%[4]。以MLPA、Real-time PCR為代表的基因定量檢測技術雖然可以明確和基因的拷貝數,但是無法區分2+0基因型和正常的1+1基因型,需要輔以其他檢測技術綜合分析。Chen等[5]早在1999年就利用基因熒光定量結合單倍型分析的方法證實了2+0基因型的存在。2000年,Yan等[6]通過將人類細胞與小鼠細胞融合后進行選擇性培養,可以分離出單個人類染色體。2001年Mailman等[7]利用上述技術成功檢測出了2+0基因型,但是這項技術耗時且費力。近來也有報道利用單精子測序的方法輔助明確男性個體2+0基因型,但不適于女性個體[8]。因此,就目前而言,基因定量技術已經成熟,對于疑似2+0基因型個體,可以優先檢測其雙親基因的拷貝數。當雙親一方為1+0攜帶者,另一方基因的拷貝數大于等于3則高度提示疑似個體為2+0基因型。
因為基因和基因的拷貝數變異很大程度上源自親代遺傳,并可以穩定地傳遞下去[9],所以2+0基因型攜帶者在人群中也會穩定存在(#8家系先證者姐姐)。而常規的檢測方法不能直接確定2+0基因型,那么發現與重復等位基因連鎖的單倍型可以有效提示2+0基因型的存在。Luo等[2]報道了在Ashkenazi猶太人中,由多態位點g.27134T> G和g.27706-27707delAT構成的單倍型僅存在于攜帶重復等位基因的個體中(拷貝數大于等于3),而不存在于對照個體(拷貝數為2)。提示這兩個多態位點可能與重復等位基因呈現連鎖不平衡。那么在一個基因拷貝數為2的個體中檢測到這兩個多態變異時,則高度提示這2個拷貝的基因位于同一條染色體,該個體為2+0攜帶者的風險增加。這一發現使得Ashkenazi猶太人群中SMA攜帶者檢出率由90%提升至94%。但也并不是所有的2+0基因型個體都可以檢測到這兩個多態變異的存在。因此在特定人群中,當攜帶2拷貝基因的個體不存在g.27134T>G或g.27706_27707delAT多態變異中的任何一個,尚不能排除2+0攜帶者的可能,但是可以降低該個體為攜帶者的風險。
最近研究顯示,非洲人群中攜帶3拷貝和4拷貝基因的個體所占比例分別為41.4%(373/902)和13.4% (121/902),顯著高于東亞人群的5.6% (33/593)和0% (0/593)[10]。而在拷貝數同樣為3或4的個體中,非洲人群g.27134T>G的檢出率為86.4% (427/494),顯著高于東亞人群的3.0% (1/33)。以上結果提示,基因拷貝數的分布以及g.27134T>G多態位點變異具有種族特異性。中國人群中攜帶3拷貝和4拷貝基因的個體所占比例分別為7.0% (1434/20403)和0.3% (60/20403)[11],尚無多態位點g.27134T>G和g.27706_27707delAT的相關數據。
本研究篩查了44例家系成員和204例攜帶不同基因拷貝數的對照樣本,僅在對照組中發現1例基因拷貝數為3的個體同時攜帶g.27134T>G和g.27706_27707delAT多態變異。因此,g.27134T>G和g.27706_27707delAT在攜帶3拷貝基因個體中的檢出率均為6.3% (1/16),g.27134T>G的檢出率顯著低于非洲人群(84.5%)[10]和西班牙人群(11.8%)[12],略高于東亞人群(3.0%)[10],g.27706_27707delAT的檢出率顯著低于西班牙人群(11.8%)[12]。而本研究在10例確定2+0基因型和18例疑似2+0基因型個體中均未發現這兩個多態變異。以上結果提示多態位點g.27134T>G和g.27706_ 27707delAT在中國人群中的比例可能相對較低,對于2+0基因型的提示作用尚需進一步擴大樣本進行驗證。同時,也需要尋找中國人群特異性的單倍型或多態位點,以提升2+0攜帶者的檢出率。但是,鑒于我國為多民族國家,是否存在普適的單倍型或多態位點還有待考究。
綜上所述,本研究通過多代系基因定量結合STR連鎖分析確定了純合缺失SMA家系中10例2+0基因型個體,為家系遺傳病的診斷提供了更為精準的遺傳咨詢。對于疑似2+0基因型個體,首先推薦檢測其雙親基因的拷貝數。此外,本研究結果也表明g.27134T>G和g.27706_27707delAT多態位點可能與中國人群2+0基因型個體的關聯度較低,尚需進一步探尋中國人群特異性的分子標記以提高2+0基因型攜帶者的檢出率。
[1] Zhu SY, XiongF, Chen YJ, Yan TZ, Zeng J, Li L, Zhang YN, Chen WQ, Bao XH, Zhang C, Xu XM. Molecularcharacterization of SMN copy number derived fromcarrier screening and from core families with SMA in a Chinesepopulation., 2010, 18: 978–984.
[2] Luo MJ, Liu L, Peter I, Zhu J, Scott SA, Zhao GP, Eversley C, Kornreich R, Desnick RJ, Edelmann L. An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan-ethnic carrier screening for spinal muscular atrophy., 2014, 16(2): 149–156.
[3] Verhaart IEC, Robertson A, Wilson IJ, Aartsma-Rus A, Cameron S, Jones CC, Cook SF, Lochmüller H. Prevalence, incidence and carrier frequency of 5q-linked spinal muscular atrophy - a literature review., 2017, 12(1): 124.
[4] McAndrew PE, Parsons DW, Simard LR, Rochette C, Ray PN,Mendell JR, Prior TW, Burghes AH. Identification ofproximal spinal muscular atrophy carriers and patients byanalysis of SMNT and SMNC gene copy number., 1997, 60(6): 1411–1422.
[5] Chen KL, Wang YL, Rennert H, Joshi I, Mills JK, Leonard DG,Wilson RB. Duplications and de novo deletions of the SMNt gene demonstrated by fluorescence-based carrier testingfor spinal muscular atrophy., 1999, 85(5): 463–469.
[6] Yan H, Papadopoulos N, Marra G, Perrera C, Jiricny J, BolandCR, Lynch HT, Chadwick RB, de la Chapelle A, Berg K, Eshleman JR, Yuan W, Markowitz S, LakenSJ, Lengauer C, Kinzler KW, Vogelstein B. Conversion of diploidy to haploidy., 2000, 403(6771): 723–724.
[7] Mailman MD, Hemingway T, Darsey RL, Glasure CE, Huang Y, Chadwick RB, Heinz JW, Papp AC, Snyder PJ, Sedra MS, Schafer RW, Abuelo DN, Reich EW, Theil KS, Burghes AH, de la Chapelle A, Prior TW. Hybrids monosomal for human chromosome 5 reveal the presence of a spinal muscular atrophy (SMA) carrier with two SMN1 copies on one chromosome., 2001, 108(2): 109–115.
[8] Burlet P, Gigarel N, Magen M, Drunat S, Benachi A, Hesters L, Munnich A, Bonnefont JP, Steffann J. Single- sperm analysis for recurrence risk assessment of spinal muscular atrophy., 2010, 18(4): 505–508.
[9] Cao YY, Qu YJ, Bai JL, Cheng MM, Jin YW, Wang H, Song F. Transmission characteristics of SMN from 227 spinal muscular atrophy core families in China., 2020, 65(5): 469–473.
[10] Chen X, Sanchis-Juan A, French CE, Connell AJ, Delon I, Kingsbury Z, Chawla A, Halpern AL, Taft RJ, BioResource N, Bentley DR, Butchbach MER, Raymond FL, Eberle MA. Spinal muscular atrophy diagnosis and carrier screening from genome sequencing data., 2020, 22(5): 945–953.
[11] Zhao SM, Wang WY, Wang YS, Han R, Fan CN, Ni PX, Guo FY, Zeng FW, Yang QN, Yang Y, Sun Y, Zhang XH, Chen Y, Zhu BS, Cai WW, Chen S, Cai R, Guo XL, Zhang CL, Zhou YQ, Huang SD, Liu YH, Chen BY, Yan SH, Chen YJ, Ding HM, Shang X, Xu XM, Sun J, Peng ZY. NGS-based spinal muscular atrophy carrier screening of 10,585diverse couples in China: a pan-ethnic study., 2020, doi: 10.1038/s41431-020-00714-8.
[12] Alías L, Bernal S, Calucho M, Martínez E, March F, Gallano P, Fuentes-Prior P, Abuli A, Serra-Juhe C, Tizzano EF. Utility of two SMN1 variants to improve spinal muscular atrophy carrier diagnosis and genetic counselling., 2018, 26(10): 1554–1557.
Familial study of spinal muscular atrophy carriers with(2+0) genotype
Yanyan Cao, Miaomiao Cheng, Fang Song, Yujin Qu, Jinli Bai, YuweiJin, Hong Wang
Spinal muscular atrophy (SMA) is a common childhood neuromuscular disease inherited in an autosomal recessive pattern. The majority of SMA patients have a homozygous deletion of survival motor neuron 1 () gene. As a special SMA carrier, the (2+0) genotype ofposes a great challenge for carrier screening and family genetic counseling. A previous study showed that polymorphisms of g.27134 T>G and g.27706_27707delAT had a predictive effect on (2+0) carriers in the Ashkenazi Jewish population. To further explore whether these two polymorphisms are specific to the Chinese population, the present study recruited 44 family members and 204 controls with knowncopy number. These 44 family members were from nine unrelated SMA families withhomozygous deletion, and one of the proband parents was suspected to be a (2+0) carrier. Multiplex ligation-dependent probe amplification (MLPA) and short tandem repeat (STR) linkage analyses were used to determine the (2+0) genotype and polymorphism screening. Finally, by analyzing thecopies and haplotype from three generations of family members and two generations of multi-child families, ten individuals in nine families were confirmed as (2+0) carriers. Moreover, only one individual with three copies ofcarried the two polymorphisms of g.27134 T>G and g.27706_27707delAT. Therefore, we provided precise genetic counseling for these SMA families after confirming the (2+0) carriers. The association between the polymorphisms of g.27134T>G and g.27706_27707delAT and Chinese (2+0) carriers might be weak. Hence, it is necessary to find specific polymorphisms in the Chinese population to improve the detection rate of (2+0) carriers.
spinal muscular atrophy;;(2+0) genotype; STR linkage analysis; polymorphism
2020-10-17;
2020-12-11
國家重點研發計劃項目(編號:2016YFC0901505),中國醫學科學院醫學與健康科技創新工程項目(編號:2016-I2M-1-008),中國博士后科學基金項目(編號:2018M630108),國家自然科學基金項目(編號:81500979)和北京市自然科學基金項目(編號:5163028)資助[Supported by the National Key Research and Development Program of China (No.2016YFC0901505), and CAMS Initiative for Innovative Medicine (No. 2016-I2M- 1-008), the China Postdoctoral Science Foundation (No.2018M630108), National Natural Science Foundation of China (No. 81500979), and Beijing Natural Science Foundation (No. 5163028)]
曹延延,博士,副研究員,研究方向:兒童罕見病的遺傳基礎。E-mail: caoyanyan@bjmu.edu.cn
宋昉,碩士,研究員,研究方向:兒童罕見病機制研究。E-mail: songf_558@263.net
10.16288/j.yczz.20-319
2020/12/28 16:51:11
URI: https://kns.cnki.net/kcms/detail/11.1913.R.20201225.1802.004.html
(責任編委: 夏昆)
