基于指紋圖譜結合多模式化學計量學方法評價補骨脂藥材質量
2021-02-25 09:41:44王玉勤范國榮
中草藥 2021年4期
關鍵詞:質量
王玉勤,范國榮, 2*
基于指紋圖譜結合多模式化學計量學方法評價補骨脂藥材質量
王玉勤1,范國榮1, 2*
1. 上海交通大學藥學院,上海 200240 2. 上海交通大學附屬第一人民醫院臨床藥學科,上海 200080
采用指紋圖譜并結合多種化學計量方法,篩選特征化學成分,區分不同來源的藥材,綜合評價中藥的質量。采用HPLC法建立補骨脂藥材指紋圖譜,相似性分析(similarity analysis,SA)、聚類分析(hierarchical cluster analysis,HCA)、主成分分析(principal component analysis,PCA)化學計量方法進行分析。色譜柱為Waters X Select HSS T3 xp(150 mm×2.1 mm,2.5 μm),流動相為乙腈(A)-水(B),梯度洗脫,0~10 min,5%~10% A;10~15 min,10%~35%A;15~25 min,35%~60%A;25~35 min,60%~85%A;35~50 min,85%~95%A;體積流量0.3 mL/min,檢測波長為246 nm,柱溫為30 ℃,進樣量為1 μL。以補骨脂素為參照,繪制11批樣品的HPLC圖譜,采用《中藥色譜指紋圖譜相似度評價軟件(2012年版)》進行相似性評價分析,確定共有峰,并采用SIMCA 13.0、SPSS 22.0軟件進行HCA和PCA。11批補骨脂樣品的HPLC圖譜有12個共有峰,根據相似度均在是否大于0.95,可將11批次藥材分為2類;采用HCA的2種計算方法,分類結果基本一致,說明樣品的一致性良好;3個主成分因子的累積方差貢獻率為94.524%,以S8樣品的主成分因子綜合得分最高、整體質量最好。所建HPLC指紋圖譜的相似度分析及聚類分析和主成分分析方法結果可為補骨脂藥材的質量控制提供參考。
補骨脂;高效液相色譜法;相似度分析;聚類分析;主成分分析
補骨脂為豆科植物補骨脂L. 的干燥成熟果實。具有溫腎助陽、納氣平喘、溫脾止瀉的功效[1]。化學研究表明補骨脂中的主要成分為香豆素類和黃酮類以及酚類化合物[2]。補骨脂素、異補骨脂素、補骨脂異黃酮、補骨脂乙素、補骨脂酚為補骨脂的主要有效成分,其中補骨脂素和異補骨脂素具有具有抗腫瘤、抗白血病、促骨形成(擬雌激素)、緩解抑郁癥等藥理作用,是評價補骨脂藥材質量的主要指標,因此對補骨脂及其制劑的含量測定多以補骨脂素和異補骨脂素這2種成分為質量控制指標[3]。其含量測定方法有薄層掃描法、氣相色譜法、高效液相色譜法,但僅采用單一藥味的顯微及薄層色譜鑒別以及補骨脂素一種成分的含量測定來控制產品的質量,無法全面反映產品的質量。鑒于此,筆者通過檢測11批補骨脂樣品,建立其高效液相色譜(HPLC)指紋圖譜[4],并結合相似度分析、聚類分析、主成分分析對其質量進行全面評價,旨在為補骨脂藥材的質量控制提供參考。
1 材料與儀器
1.1 材料
11批補骨脂藥材購于云南、河南、江蘇等地,經范國榮教授鑒定為豆科植物補骨脂L.的干燥成熟果實,具體信息見表1。補骨脂素(批號1018444-F2290010,質量分數大于98%)、異補骨脂素(批號1017007-HO150006,質量分數大于98%)購自上海安譜實驗科技有限公司;補骨脂異黃酮(批號112021-201601,質量分數大于98%);補骨脂乙素(批號MB7009,質量分數大于98%);補骨脂酚(批號MB6546,質量分數大于95%);甲醇、乙腈(色譜純,Merck(Darmstadt公司,德國),其他試劑均為分析純,超純化水(Milli-Q系統)。

表1 樣品信息
1.2 儀器
Thermo U-3000超高效液相色譜儀,配備自動進樣器、DAD檢測器、變色龍7.0工作站(美國Thermo Fishe Scientific公司);旋轉蒸發儀(上海信科科技有限公司);DionexTMASETM350加速溶劑萃取儀(美國Thermo Fishe Scientific公司);Millipore(Bedford公司,MA,美國);Eppendorf 5804R型高速冷凍離心機(Eppendorf公司,德國);Scientific Industries Vortex-Genic 2型渦旋混合器(Scientific Industries公司,美國);SK5200H超聲儀(上海科導超聲儀器有限公司);Sartorius CPA 225D十萬分之一電子天平(賽多利斯公司,德國);梅特勒托利多AL104-01萬分之一電子天平(梅特勒托利多公司,德國);DJ-O4B型中藥粉粹機(上海淀久中藥機械有限公司公司)。
2 方法與結果
2.1 溶液的制備
2.1.1 對照品溶液的制備 分別精密稱取補骨脂素10.02 mg、異補骨脂素10.01 mg、補骨脂異黃酮17.50 mg、補骨脂乙素5.20 mg、補骨脂酚6.05 mg對照品適量,分別置于25 mL量瓶中,加甲醇制得質量濃度分別為400.8、400.4、700.0、208.0、242.0 μg/mL的對照品溶液。
2.1.2 供試品溶液的制備 將11批次干燥補骨脂藥材分別放入粉碎機中粉碎1 min,過2號篩、40目的粗篩,稱取補骨脂粗粉約1.000 g,與硅藻1∶1,充分混勻后,置于樣品池中進行ASE萃取,收集萃取液,減壓回收溶劑,加入適量甲醇溶解,定容至25 mL量瓶中,精密吸取1 mL加50%甲醇定容至10 mL量瓶中,取1 mL供試品溶液,13 000 r/min 離心10 min,重復離心2次,上清液作為供試品溶液進HPLC分析。
2.2 色譜條件
色譜柱為Waters X Select HSS T3 xp(150 mm×2.1 mm,2.5 μm),流動相為乙腈(A)-水(B),洗脫梯度:0~10 min,5%~10% A;10~15 min,10%~35%A;15~25 min,35%~60%A;25~35 min,60%~85%A;35~50 min,85%~95%A,體積流量0.3 mL/min,檢測波長為246 nm,柱溫為30 ℃,進樣量為1 μL。混合對照品和樣品色譜圖見圖1。

1-補骨脂素 2-異補骨脂素 6-補骨脂異黃酮 9-補骨脂乙素 12-補骨脂酚
2.3 方法學考察
2.3.1 線性關系考察 分別精密移取“2.1.1”項對照品母液適量,用50%甲醇逐級稀釋成補骨脂素質量濃度分別為269.123、134.562、67.281、33.640、16.820、8.410、4.205 μmol/L;異補骨脂素質量濃度分別為268.850、134.425、67.212、33.606、16.803、8.401、4.200 μmol/L;補骨脂異黃酮質量濃度分別為182.073、91.036、45.518、22.759、11.380、5.690、2.845 μmol/L;補骨脂乙素質量濃度分別為160.311、80.155、40.078、20.039、10.019、5.010、2.505 μmol/L;補骨脂酚質量濃度分別為235.978、117.989、58.994、29.497、14.749、7.374、3.687 μmol/L;進樣10 μL測定。以對照品質量濃度()對峰面積()進行線性回歸,補骨脂素的線性回歸方程=0.493 9+0.068 7,2=0.999 9;異補骨脂素的線性回歸方程:=0.517 1+0.111 9,2=0.999 9;補骨脂異黃酮的線性回歸方程=0.181 5+0.022,2=0.999 9;補骨脂乙素的線性回歸方程=0.058 6-0.006 4,2=1.000 0;補骨脂酚的線性回歸方程:=0.038 7-0.004 9,2=1.000 0;結果表明,5個化合物在各自的濃度范圍內線性關系良好,且2均在0.999 9以上。
2.3.2 精密度試驗 將補骨脂供試品溶液(批號180101)連續進樣6次,記錄各主要峰的峰面積,5個特征峰相對峰面積RSD值分別為0.552%、0.570%、0.735%、0.916%、0.486%。
2.3.3 穩定性試驗 取同一補骨脂試品溶液(批號180101),分別于室溫下放置0、2、4、8、12、24 h進樣分析,記錄各主要峰的峰面積。結果,5個特征峰相對峰面積RSD值分別為1.000%、0.885%、3.566%、1.519%、1.154%。
2.3.4 重復性試驗 取同一批補骨脂樣品(批號1801011),按“2.1.2”項下方法平行制備6份,進樣分析,記錄各主要峰的峰面積。結果表明,5個特征峰相對峰面積RSD值為0.925%、1.035%、3.558%、1.270%、0.992%。
2.3.5 加樣回收率試驗 精密稱取6份已測定的補骨脂藥材(批號1801011)粉末適量,分別加入等量的的5種對照品,按照“2.1.2”項下供試品溶液的制備方法制備供試品溶液,依法測定并計算加樣回收率,補骨脂素、異補骨脂素、補骨脂異黃酮、補骨脂乙素、補骨脂酚的平均回收率為92.32%、91.80%、102.99%、104.26%、107.79%,RSD%分別為3.99%、1.98%、1.38%、1.09%、1.03%。
2.4 補骨脂藥材中特征性成分的定量測定
分別取不同來源的11批補骨脂藥材精密稱定,根據“2.2”項色譜條件,測定補骨脂素、異補骨脂素、補骨脂異黃酮、補骨脂乙素、補骨脂酚的含量見表2。
2.5 指紋圖譜的建立
2.5.1 精密度試驗 取補骨脂藥材(S1)供試品溶液,按“2.2”項下確定的色譜條件下連續進樣6次,檢測指紋圖譜,以補骨脂酚色譜峰為參照峰(S),計算各共有指紋峰與S峰的相對保留時間和相對峰面積RSD值均小于3.0%,表明儀器精密度良好。
2.5.2 重復性試驗 取同一批次的補骨脂藥材粉末(S1)共計6份,按“2.1.2”項下確定的供試品溶液制備方法制備6份供試品溶液,分別進樣測定,檢測指紋圖譜,以補骨脂酚色譜峰為參照峰(S),計算各共有指紋峰與S峰的相對保留時間和相對峰面積RSD值均小于3.0%,表明該方法重復性良好。
2.5.3 穩定性試驗 取補骨脂藥材(S1)供試品溶液,分別在0、2、4、6、8、12、24 h進樣分析,檢測指紋圖譜,以補骨脂酚色譜峰為參照峰(S),計算各共有指紋峰與S峰的相對保留時間和相對峰面積RSD值均小于3.0%,說明供試品溶液在24 h內穩定性良好。
2.5.4 指紋圖譜的建立及相似度評價 取11批補骨脂樣品(S1~S11),按照“2.1.2”項下方法制備供試品溶液,再按“2.2”項下色譜條件進樣測定,將得到的色譜圖導入《中藥色譜指紋圖譜相似度評價系統(2012年版)》,以S1為參考圖譜,時間窗寬度設為0.1 min,手動匹配11批指紋圖譜(采用多點校正),建立補骨脂HPLC指紋圖譜的共有模式(平均值法)[5-6]。根據匹配結果確定12個共有峰,并生成對照指紋圖譜,計算出各批次樣品的指紋圖譜相似度。在所有共有峰中,12號峰對照品易得,并且該成分色譜峰面積最大,分離度及對稱性均良好,確定為參照峰(S)。相似度測定結果分別見表3。S1~S11指紋圖譜疊加圖譜和對照指紋圖譜見圖2、3。

表2 不同產地補骨脂中藥材中5種主要成分的含量

圖2 11批樣品的HPLC疊加指紋圖譜

1-補骨脂素 2-異補骨脂素 6-補骨脂異黃酮 9-補骨脂乙素 12-補骨脂酚
2.5.5 指紋圖譜中共有峰指認 通過與對照品圖譜(圖3)比對可以指認其中的5個色譜峰,可作為特征峰,分別是補骨脂素(1號峰)、異補骨脂素(2號峰)、補骨脂異黃酮(6號峰)、補骨脂乙素(9號峰)、補骨脂酚(12號峰)。
2.6 相似度分析
相似性分析(similarity analysis,SA)[7-8],精確計算原始數據的相關系數,對樣本的相似性和差異性進行分類,并對原始數據進行識別,用相關系數評價它們的相似度見表3,一般來說,相似度越接近于1,2種色譜圖越接近,基于11批次補骨脂相似度分析的結果,將相似性系數大于0.95的分為1組,其余的分為另1組;即S1、S4、S5、S7、S11為1類;S2、S3、S6、S8、S9、S10為1類。
2.7 聚類分析
聚類分析(hierarchical cluster analysis,HCA)是一組將研究對象分為相對同質的群組(clusters)的統計分析技術,也稱分類分析(classification analysis)或數值分類,實質是在多維空間中將樣本看作不同的點,用不同的測量方法計算樣本點之間的距離,并建立分類基[9-10]。
以11批樣品的12個共有峰的相對峰面積為原始數據,采用SPSS 22.0軟件,以Ward最小方差法以及組間平均連接的系統聚類法結合歐氏距離()為測度進行分析,結果見圖4。由圖4可知,用Ward最小方差法,=25時,11批樣品可聚為1類;=5時,后一類又可聚為4類,即S2、S5、S6、S7聚為1類,S3、S9聚為1類,S1、S4、S10聚為1類,S11為1類;用系統聚類法,=25時,11批樣品可聚為1類;=5時,后1類又可聚為2類,即S5、S7、S2、S6聚為一類,S3、S9聚為1類。
2.8 主成分分析 (principal component analysis,PCA)
PCA作為一種數據簡約技術,即無監督數據降維。通過建模方法將多維數據標準化,消除變量之間在數量級上的差異,計算標準化數據的相關矩陣并計算相關矩陣的特征向量、特征值和累積貢獻率,確定主成分的個數,計算每個樣品的綜合函數得分,以該得分進行排序評價,并能提供一個可視化的分布視圖[11-12]。
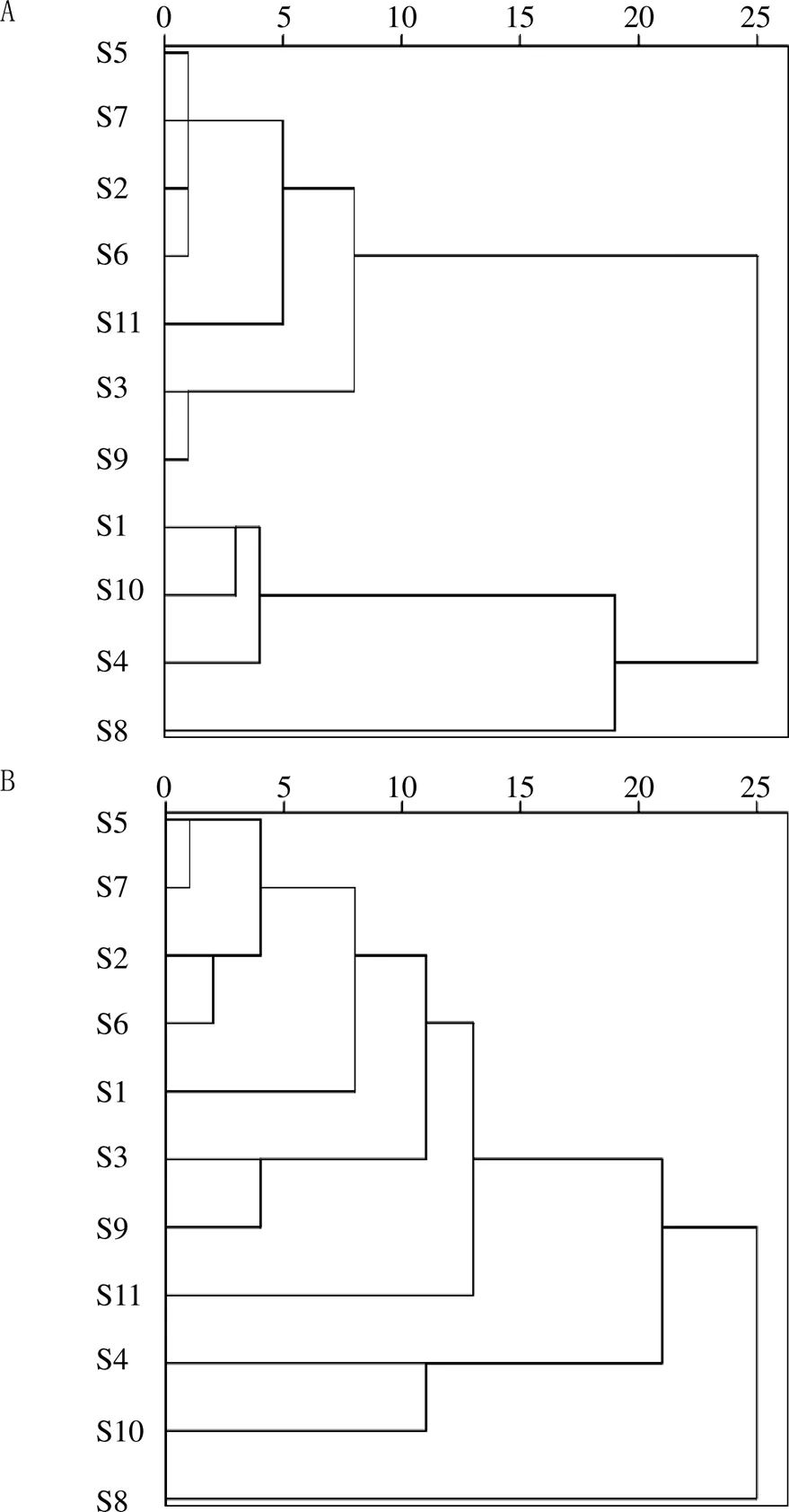
A-Ward最小方差法計算平方歐氏距離 B-歐幾里得法計算平方歐氏距離
以各共有峰的相對峰面積為指標,將11批補骨脂12個共有峰的相對峰面積進行標準化處理,采用SPSS 22.0軟件和SIMCA 13.0軟件進行PCA,得到12種共有成分在11批次補骨脂樣品中的得分圖和裝載圖,見圖5,表明不同產地的補骨脂藥材的化學成分指紋圖譜有明顯的差別。在載荷圖中選取距離原點較遠的幾個點,得到主成分1中變量的權重值,權重值越大表明該成分在決定樣品區分中的作用越大,其中前4名變量的權重值分別為0.58、0.90、0.14和0.11,它們的保留時間分別為21.81、22.19、27.82和30.22 min,分別為指紋圖譜中的1、2、6和9號色譜峰。
用SPSS 22.0軟件計算其特征值和方差貢獻率,以特征值>1為標準,得到前3個因子的特征值分別為7.001、3155、1.186,是影響補骨脂質量評價的主要因子[13]。結合表4,前3個主成分因子的累積方差貢獻率為94.524%。選擇提取該3個主成分因子對11批補骨脂樣品進行評分,以方差貢獻率為分配系數,線性組合3個主成分因子,計算各批樣品的主成分因子得分及綜合得分,并對綜合得分進行排序[14],結果見表5,綜合得分由高到低的批次依次為S8、S3、S9、S7、S2、S5、S10、S1、S11、S4。
3 討論
中藥指紋圖譜是一種多指標質量控制模式,可通過表征中藥復雜成分及其內在質量的關系來較全面地反映所含化學成分的種類和數量,從而反映藥品的質量,在中藥制劑的質量評價中應用廣泛[15]。本研究通過查閱文獻資料[16-17]在制備測定補骨脂HPLC供試品溶液時,考察了提取工藝以及色譜柱、流動相、波長、溫度等因素的選擇,確定最佳提取條件和色譜條件。
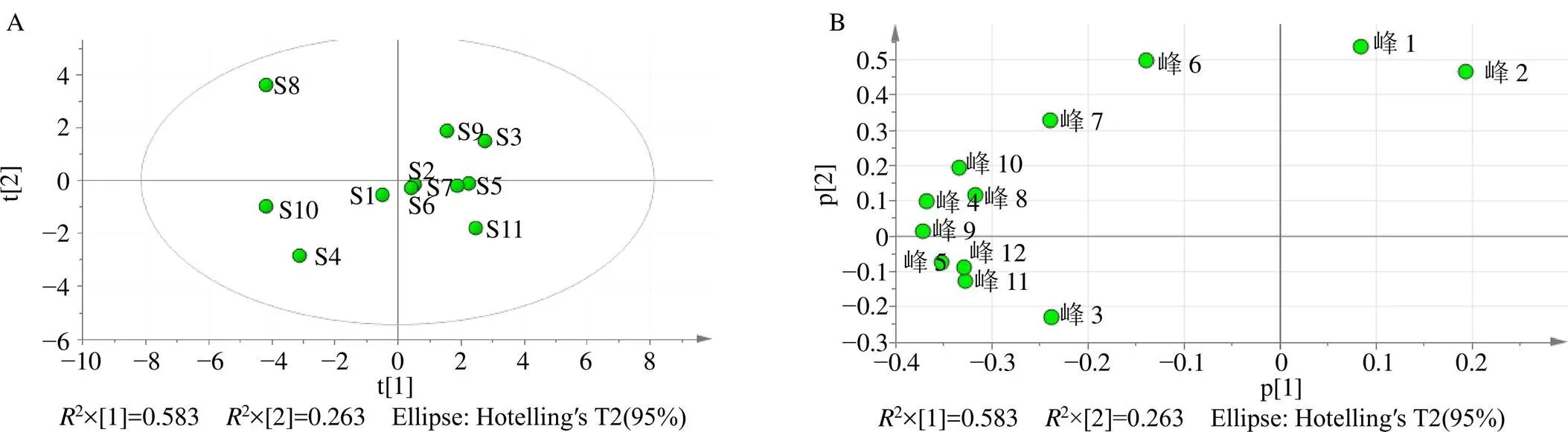
圖5 11批次樣品的主成分分析PCA評分圖(A) 和12種共有成分的PCA裝載圖(B)

表4 3個主成分因子的特征值和方差貢獻率

表5 11批補骨脂樣品主成分因子得分及排序
本實驗所建立的補骨脂HPLC指紋圖譜,可反映補骨脂的整體性和特異性信息,在12個共有峰中,通過與對照品HPLC圖譜比對指認出5個成分。SA選擇成分的平均值進行分析,精確計算原始數據的相關系數,對樣本的相似性和差異性進行分類,將樣品的共有峰作為特征性成分進行定量的計算標記;HCA和PCA則是選擇樣品中的相似對象進行分類分析,通過對象傾向找出潛在的聚類,將整個色譜圖轉化為統計結構,將不同來源的樣品分成不同的簇和類別。本研究具備了定性、鑒別和定量測定的雙重作用,提高了中藥材的內在質量控制水平,為不同化學計量方法與質量評價的比較提供了一種新的綜合策略,對中藥材的質量控制提供了準確性和可靠性的有價值的參考依據。
利益沖突 所有作者均聲明不存在利益沖突
[1] 中國藥典 [S]. 一部. 2015: 187.
[2] 周曉英, 黃曉婧, 周世玉, 等. 《中國藥典》補骨脂項下含量測定方法的探討 [J]. 中國藥品標準, 2015, 16(1): 3-4.
[3] 趙婷, 朱汀瀅, 吳斌. 補骨脂中補骨脂素和異補骨脂素測定 [J]. 中成藥, 2015, 37(5): 1036-1040.
[4] Zhang Y M, Chen Z Y, Xu X K,. Rapid separation and simultaneous quantitative determination of 13 constituents inby a single marker using high-performance liquid chromatography with diode array detection [J]., 2017, 40(21): 4191-4202.
[5] 沈曉宇, 劉雪松, 畢宇安, 等. 鹽補骨脂標準湯劑質量評價體系的建立 [J]. 中草藥, 2018, 49(1): 100-108.
[6] 伍紅年, 譚詩涵, 王元清, 等. 白三七及近源種藥材指紋圖譜與識別模式的構建及其應用研究 [J]. 中草藥, 2019, 50(1): 217-224.
[7] 龍立梅, 宋沙沙, 曹學麗. 基于香氣成分氣相色譜-質譜指紋圖譜的判別分析和相似度評價用于綠茶等級差異研究 [J]. 色譜, 2019, 37(3): 325-330.
[8] Yang G, Zhao X, Fan G. A modification on the vector cosine algorithm of similarity analysis for improved discriminative capacity and its application to the quality control of[J]., 2017, 1518: 34-45.
[9] Hans-Friedrich K?hn, Lawrence J H. Hierarchical cluster Analysis [M]. Wiley StatsRef: Statistics Reference Online, 2015: 236.
[10] 林林, 于鳳蕊, 徐麗華, 等. 女金丸HPLC指紋圖譜建立及聚類分析和主成分分析 [J]. 中國藥房, 2019, 30(10): 1339-1343.
[11] Gao F Y, Xu Z H, Wang W Z,. A comprehensive strategy using chromatographic profiles combined with chemometric methods: Application to quality control ofSieb. et Zucc [J]., 2016, 1466: 67-75.
[12] Granato D, Santos J S, Escher G B,. Use of principal component analysis (PCA) and hierarchical cluster analysis (HCA) for multivariate association between bioactive compounds and functional properties in foods: A critical perspective [J]., 2018, 72: 83-90.
[13] 蔡曉洋, 張思荻, 曾俊, 等. 基于主成分分析和聚類分析的梔子種質資源評價 [J]. 中國實驗方劑學雜志, 2017, 23(14): 30-37.
[14] 王洪蘭, 姚衛峰, 朱丹妮, 等. 不同來源瓜子金藥材的HPLC-DAD-ELSD指紋圖譜和主成分分析 [J]. 中國天然藥物, 2010, 8(5): 343-348.
[15] 刁嘉茵, 徐燦, 王淑美, 等. 中藥指紋圖譜與藥效相關性研究進展 [J]. 藥學研究, 2018, 37(3): 165-168.
[16] 李凱, 魯亞奇, 周寧. 補骨脂中補骨脂素和異補骨脂素提取工藝研究 [J]. 中醫學報, 2018, 33(9): 1716-1720.
[17] Zhu L, Wang Z, Zhai X,. Simultaneous quantitative determination of 13 active components in the traditional Chinese medicinal preparation Suanzaoren oral liquid by HPLC coupled with diode array detection and evaporative light scattering detection [J]., 2017, 40(11): 2320-2325.
Quality evaluation ofby using chromatographic profiles combined with chemometric methods
WANG Yu-qin1, FAN Guo-rong1, 2
1. School of Pharmacy, Shanghai Jiao Tong University, Shanghai 200240, China 2. Department of Clinical Pharmacy, Shanghai General Hospital, Shanghai Jiao Tong University, Shanghai 200080, China
To distinguish the medicinal materials from different sources and evaluate the quality of Chinese herbal medicines by using chromatographic profiles combined with chemometric methods.HPLC, similarity analysis (SA), cluster analysis (HCA) and principal component analysis (PCA) were used to analyze the original data. The chromatographic column was Waters X Select HSS T3 xp (150 mm×2.1 mm, 2.5 μm), the mobile phase was acetonitrile-water (gradient elution), the flow rate was 0.3 mL/min, the detection wavelength was 246 nm, the column temperature was 30 ℃, and the injection volume was 1 μL. HPLC profiles of 11 batches of samples were drawn using psoralen as the reference. Similarity evaluation software of Chinese traditional medicine chromatographic fingerprint (2012 edition) was used for similarity evaluation and analysis to determine the common peaks, and hierarchical cluster analysis and principal component analysis were conducted by SIMCA 13.0 and SPSS 22.0 software.There were 12 common peaks in the fingerprint of 11 batches of. samples. According to whether the similarity index was > 0.95 or not, 11 batches of medicinal materials could be divided into two categories. Two methods of hierarchical cluster analysis were adopted, and the classification results were basically consistent, indicating the good consistency of samples. The cumulative variance contribution rate of the three principal component factors was 94.524%, and the S8 sample had the highest comprehensive score of principal component factors and the best overall quality.The results of similarity analysis, cluster analysis and principal component analysis of HPLC fingerprint can provide reference for quality control ofsamples.
L.; high performance liquid chromatography; similarity analysis; hierarchical cluster analysis; principal component analysis
R286.2
A
0253 - 2670(2021)04 - 1143 - 07
10.7501/j.issn.0253-2670.2021.04.028
2020-06-03
上海市衛生和計劃生育委員會中醫藥科研專項(2016ZJP003)
王玉勤(1995—),女,漢族,山東菏澤人,碩士研究生。Tel: 15201973182 E-mail: 18865737427@163.com
范國榮(1965—),男,漢族,浙江慈溪人,教授,博士研究生導師,主要從事臨床藥學和臨床藥代動力學研究。Tel: 13301777863 E-mail: guorfan@163.com
[責任編輯 時圣明]
猜你喜歡
中學生數理化·中考版(2022年10期)2022-11-10 09:37:42
中學生數理化·八年級物理人教版(2022年12期)2022-02-14 07:08:42
中學生數理化·八年級物理人教版(2021年12期)2021-12-31 03:23:08
中學生數理化·中考版(2020年10期)2020-11-27 01:59:48
中國生殖健康(2019年2期)2019-08-23 08:12:08
石油化工建設(2018年6期)2018-04-22 03:16:54
產品可靠性報告(2017年7期)2017-09-05 09:49:12
中學生數理化·八年級物理人教版(2017年12期)2017-04-18 12:59:38
汽車觀察(2016年3期)2016-02-28 13:16:26
民生周刊(2014年7期)2014-03-28 01:30:54
