液相體系Ru-Co3O4 催化CO2 加氫制甲烷過程中的溶劑效應研究
2021-02-24 06:15:32宋英健崔曉靜鄧天昇秦張峰樊衛斌
燃料化學學報 2021年2期
關鍵詞:催化劑
宋英健 ,崔曉靜 ,鄧天昇 ,秦張峰 ,樊衛斌,*
(1.中國科學院山西煤炭化學研究所 煤轉化國家重點實驗室,山西太原 030001;2.中國科學院大學,北京 100049;3.太原工業學院 化學與化工系,山西 太原 030008)
含碳資源的利用會產生大量二氧化碳(CO2)。這些CO2排放到大氣中,造成了一系列環境問題,如全球氣溫上升、海洋酸化等[1]。將CO2通過加氫的方式綠色高效地催化轉化為有價值的化學品,不僅具有重要的價值,而且對于能源儲存、CO2減排和可持續社會模式的建立也具有重要的意義[2]。甲烷(CH4)分子作為載能分子,可以高效地儲存氫能,而且可以利用現有天然氣基礎設施進行安全的運輸和使用,因此,將CO2催化加氫制CH4是CO2轉化利用的一條路線,受到了研究者的廣泛關注[3]。
目前,CO2催化加氫制CH4的反應過程分為氣相和液相兩種。其中,氣相加氫反應方面的研究較多,也取得了重要進展[4]。在氣相反應過程中,為了獲得較高的CO2轉化率(60%-100%),反應溫度通常較高,常在250-450 °C;同時為了抑制逆水煤氣反應(CO2+H2→CO+H2O)的活性和副產物CO 的選擇性,反應常在常壓或接近常壓的條件下進行,催化劑的甲烷時空收率往往不高[5-7]。相比之下,液相CO2加氫制CH4在較低反應溫度下(160-200 °C)就可以進行。在液相反應中,研究者多采用有機配體修飾的貴金屬均相體系為催化劑;盡管這些催化劑表現出較高的本征活性,但催化劑成本高且分離和回收利用困難等問題限制了其大規模的商業化應用[8-10]。開發負載型貴金屬多相催化劑體系有利于解決上述問題。但目前關于貴金屬多相催化劑的液相CO2加氫反應的研究報道較少,這主要是由于貴金屬多相催化劑的轉化率不高。如Dorner 等[11]研究了Co-Pt/Al2O3催化劑的液相CO2加氫活性;他們發現在220 °C、1.9 MPa、H2/CO2= 1∶1 的條件下,甲烷的選擇性可達93%,但CO2的轉化率較低,為6.8%。因此,提高貴金屬多相催化劑的轉化率是目前液相CO2加氫制CH4的挑戰之一。
在液相反應體系中,除了催化劑之外,溶劑也是重要的組成部分;溶劑對催化反應的影響規律和作用本質,即溶劑效應,是液相催化反應體系的基本科學問題之一,受到研究者的普遍關注[12]。在液相CO2加氫反應中,人們發現溶劑對反應活性影響很大。如He 等[13]采用Pt/Co3O4催化劑,深入研究了水這一溶劑在液相CO2加氫生成C2+醇類的作用本質;他們通過D2O 和13CH3OH 同位素標記實驗發現,溶劑水通過參與加氫反應,降低了反應所需的活化能,從而降低了反應所需的溫度。Filonenko 等[14]研究了負載Au 催化劑催化的液相CO2加氫制甲酸反應過程中的溶劑效應,發現甲酸的產率隨著溶劑極性增加而增加。對于液相CO2加氫制CH4反應過程,如果能深入理解溶劑效應本質,不僅有望提高反應活性,而且對設計高效的催化反應體系也具有指導意義。但目前關于液相CO2加氫制CH4反應過程中的溶劑效應,還極少見文獻報道。
鑒于此,本研究以液相CO2加氫制CH4反應為反應體系,以多相Ru-Co3O4體系為催化劑,以不同的溶劑(水、正丁醇、丁內酯、DMF、正壬烷、異辛烷、環己烷、十氫萘等)為反應體系,結合同位素標記實驗和原位紅外光譜,深入探究溶劑的作用本質。研究發現,以異辛烷和十氫萘為溶劑時,Ru-Co3O4的CO2轉化率活性最高,分別可達42%和45.6%,甲烷選擇性分別可達98.2%和97%。氘代實驗發現,這兩種溶劑促進催化劑活性的本質是它們可作為活性氫的載體,起到供氫的作用,從而促進加氫反應;相比伯碳和仲碳,叔碳具有更高的供氫能力。
1 實驗部分
1.1 催化劑的制備
實驗所用試劑如下:硝酸鈷(Co(NO3)2·6H2O,國藥集團化學試劑有限公司),碳酸鈉(Na2CO3,科密歐化學試劑有限公司),氯化釕(RuCl3,阿拉丁試劑有限公司),白炭黑(SiO2,阿拉丁試劑有限公司),氧化鋯(ZrO2,阿拉丁試劑有限公司),氧化鈰(CeO2,阿拉丁試劑有限公司),P25(TiO2,ACROS ORCANICS 公司),正丁醇(C4H10O,國藥集團化學試劑有限公司),1,4-丁內酯(C4H6O2,阿拉丁試劑有限公司),正壬烷(C9H20,杭州煉油廠),十氫萘(C10H18,阿拉丁試劑有限公司),異辛烷(C8H18,天津市福晨化學試劑廠),N,N-二甲基甲酰胺(DMF,北京北化精細化學品有限責任公司),環己烷(C6H12,阿拉丁試劑有限公司),以上試劑均為分析純。
Ru-Co3O4催化劑采用共沉淀法制備,Ru 負載量為0.96%(質量分數),與理論負載量1%相似[13]。具 體 制 備 方 法 如 下:將1.5 g 的Co(NO3)2·6H2O 和8.5 mg 的RuCl3溶解在20 g 的蒸餾水中,然后在30 °C 下20 min 內將溶液滴加到100 mL 的0.5 mol/L Na2CO3溶液中,繼續攪拌1 h;沉淀物通過離心分離得到,并用蒸餾水反復洗滌,直到硝酸銀(0.1 mol/L)溶液檢測不到白色沉淀。然后,將所得的沉淀在80 °C 干燥過夜,再在400 °C 焙燒6 h,得到Ru-Co3O4催化劑。
Ru/SiO2、Ru/ZrO2、Ru/TiO2和Ru/CeO2催化劑(Ru 的負載量為1%(質量分數))采用濕浸漬法制備。將5 g 載體加到10 mL 50 mmol/L 的RuCl3水溶液中,在室溫下攪拌2 h;所得混合物在80 °C 干燥過夜,再在300 °C 焙燒2 h。
1.2 催化劑的表征
X 射線粉末衍射(XRD)譜圖在日本Rigaku 公司的Mini Flex II X 衍射儀上獲得。儀器采用CuKα輻射(λ= 0.15406 nm),管電壓30 kV,管電流15 mA,5°-80°掃描,掃描速率為8(°)/min。
掃描電子顯微鏡(SEM)測試在日本電子株式會社的JSM-6700F 型場發射電子顯微鏡上進行。
透射電子顯微鏡(TEM)在日本電子株式會社JEM -2010 高分辨透射電子顯微鏡上采集。
核磁共振氫譜(1H-NMR)是在布魯克AV-Ⅲ400 MHz 液體核磁共振波譜儀上測得的。該液體核磁共振波譜儀配備一個5 mm BB/19F-1H/D 探頭。1H 共振頻率為400.13 MHz。
氣相色譜-質譜分析(GCMS)是在Shimadzu GCMS-QP2010Ultra 氣相色譜-質譜聯用儀(配有Rtx-5MS 毛細管柱(60 m × 0.25 mm × 0.25 μm))上進行。
原位漫反射紅外光譜圖在 Nicolet iS10 FT-IR光譜儀上采集,掃描的波長為 4000-650 cm-1,掃描64 次,分辨率為4 cm-1。在十氫萘的吸附實驗中,將一定量的催化劑粉末置于原位反應池中,以氬氣將十氫萘鼓泡帶入原位池中直至十氫萘吸附飽和,然后室溫下采集光譜,隨后再通入氬氣在室溫下對原位池進行吹掃,吹掃完畢后采集光譜;在CO2原位升溫反應實驗中,將一定量的催化劑粉末置于原位反應池中,在室溫下通入氬氣吹掃,并采集譜圖,作為背景;然后將溶劑滴加在催化劑上,通入CO2,使用加熱控溫裝置控制樣品池的溫度,采集不同溫度下的紅外光譜。
程序升溫還原(H2-TPR)在Micromeritics 公司的 AutoChemⅡ2920 型多用吸附儀上進行。以Ar 為載氣,H2+Ar(H2體積分數10%)的混合氣作為還原氣,氣體進入吸附儀前經過脫水、脫有機雜質、脫氧等多級凈化處理,熱導檢測器(TCD)監測耗氫量。樣品裝量 50 mg(粉末),首先在300 °C下用Ar(30 mL/min)吹掃30 min,隨后Ar 氣氛中冷卻至室溫,切換為還原氣,待TCD 信號穩定后,以10 °C/min 的速率從室溫升至600 °C,記錄程序升溫過程中的TCD 信號。
CO 脈沖吸附在Micromeritics 公司的 AutoChemⅡ2920 型多用吸附儀上進行。以Ar 為載氣,H2+Ar(H2體積分數10%)的混合氣作為還原氣,氣體進入儀器前經過脫水、脫有機雜質、脫氧等多級凈化處理,熱導檢測器(TCD)檢測氣體消耗量。樣品填裝量100 mg(粉末)。首先在100 °C 的還原氣氛(30 mL/min)中還原1 h;然后經Ar 吹掃1 h降溫至30 °C,恒溫條件下脈沖注入一氧化碳氣體至吸附至飽和。
1.3 催化劑性能評價和溶劑溶解度實驗
催化劑的性能評價是在30 mL 的高壓反應釜中進行(帶有聚四氟乙烯內襯)。反應前,將0.1 g的催化劑粉末在300 °C、H2/Ar 混合氣(H2和Ar 的體積比為1∶9,60 mL/min)中的管式爐中還原2 h;然后,在室溫下加入到含有5 g 溶劑的反應釜中,并將4 MPa 的H2/CO2混合氣(H2∶CO2= 3∶1,物質的量比)充入反應釜中待溫度升至200 °C 時,反應開始計時。在所有反應中,反應時間均為1 h。
待反應結束后,將反應釜放置在冰水浴中,快速冷卻至室溫;然后用2 L 的氣袋將反應后的氣體收集起來;尾氣的氣體組成采用Agilent 7890A 氣相色譜儀分析,該氣相色譜儀配有一個熱導檢測器(TCD)和兩個火焰離子化檢測器(FID),其中,H2、CO2、CO 和CH4由TCD 檢測器檢測(5A 分子篩柱,3 m × 1/16, 80/100 目),CH4和C2-6烴類由一個FID 檢測器檢測(Al2O3毛細管柱,50 m × 0.530 mm ×15.0 μm),另外一個FID 檢測器配備GS-OxyPLOT毛細管柱(10 m × 0.530 mm × 10.0 μm),用來檢測含氧類產物(如甲醇、乙醇和二甲醚等)和芳烴類產物(如苯、甲苯、二甲苯和三甲苯等)。在所有的反應中,氣相產物中均未檢測到含氧類產物和芳烴類產物。
溶解度實驗同樣是在30 mL 的高壓反應釜中進行。在25 °C 下,將12.5 mL 的溶劑加入反應釜中,然后將3 MPa 的H2充入反應釜中,密封,確保不漏氣;為了準確測得反應釜的壓力變化,采用靈敏度為1 kPa 的數字顯示壓力表來計量壓力變化;反應釜壓力不再發生變化時的值記為p1,25 °C 下溶劑的飽和蒸氣壓記為p2,氫氣溶解于溶劑中引起的壓力變化 = 3 MPa - (p1-p2)。氫氣在溶劑中的溶解度按標況下理想氣體方程計算n= Δpv/(8.314 × 298.15)。
氘代標記實驗同樣是在30 mL 的高壓反應釜中進行。將0.1 g 的Ru-Co3O4催化劑粉末在300 °C下及H2/Ar 混合氣氣氛(H2和Ar 的體積比為1∶9,60 mL/min)中的管式爐中還原2 h;隨后于室溫下加入到含有5 g 溶劑(正壬烷、十氫萘或者異辛烷)的反應釜中,然后充入D2直到壓力達到1 MPa,密封,確保不漏氣;將反應釜溫度從室溫升至200 °C,并在200 °C 下分別保持3 h(正壬烷、十氫萘)和12 h(異辛烷)。然后,將反應釜放置在冰水浴中,快速冷卻至室溫;取出溶劑進行GCMS 和核磁共振氫譜分析。
十氫萘和異辛烷的氘代實驗中,將87.1 mg 吡嗪溶解在1 mL 氘代氯仿作為內標物,每次取200 μL內標和200 μL 樣品混合進行評價。通過內標法來計算不同位置氫的氘代率。
1.4 數據處理方式
每次反應之前將反應釜浸沒在水中保證不漏氣的情況下再進行反應。
CO2轉化率和各組分選擇性的計算方法如下:

2 結果與討論
2.1 不同負載Ru 催化劑的CO2 甲烷化性能
圖1 為Ru-Co3O4催化劑的SEM 和TEM 照片,在新鮮Ru-Co3O4催化劑中,新鮮樣品Co3O4的晶粒堆疊團聚,形成較大顆粒,沒有觀察到Ru 晶粒。同時,一氧化碳脈沖化學吸附實驗[15-17]測得Ru 分散度為12.2%,說明Ru 高分散在Co3O4中。XRD 表征結果(圖2)進一步證明了這一點,新鮮樣品的XRD 譜圖與單純Co3O4的一致,沒有Ru 的特征峰,31.5°、36.8°、44.8°、59.6°和65.3°處的衍射峰分別對應Co3O4的(220)、(311)、(400)、(511)和(440)晶面[18]。

圖1 Ru-Co3O4 的SEM((a)、(b))和TEM 照片(c)Figure 1 SEM images ((a), (b)) and TEM images (c) of Ru-Co3O4
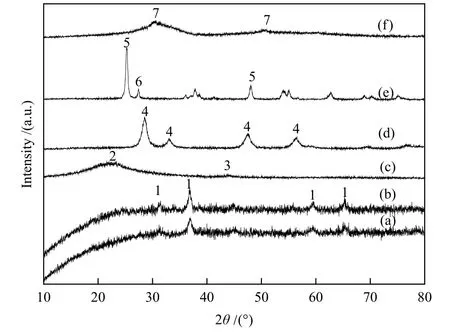
圖2 不同焙燒樣品的XRD 譜圖Figure 2 XRD patterns of calcined catalysts
在200 °C、4 MPa(H2/CO2= 3∶1)的反應條件下,反應1 h 后,Ru-Co3O4的CH4選擇性為97%,CO2轉化率為45.6%。相比較而言,Co-Pt/Al2O3,在220 °C、1.9 MPa、H2/CO2= 1∶1 的條件下,其CO2轉化率不到10%[11]。由圖2 可知,Ru 在SiO2、CeO2、ZrO2和TiO2載體上的分散度也很高,只有Ru/SiO2在44°處出現一個不明顯的包峰[19]。由表1 可知,載體對反應活性影響顯著。Ru/SiO2催化劑的CO2加氫反應活性最低,CO2轉化率僅為2.3%;Ru/CeO2催化劑的活性也較低,CO2轉化率僅為5.4%;Ru/ZrO2和Ru/TiO2催化劑的活性達到11%-15%。載體種類對產物的選擇性影響不大,不同催化劑的CH4選擇性在97%-99%。除CH4外,還有少量C2-5的烴類生成。未檢測到含氧化合物如甲醇、二甲醚、乙醇等產物。
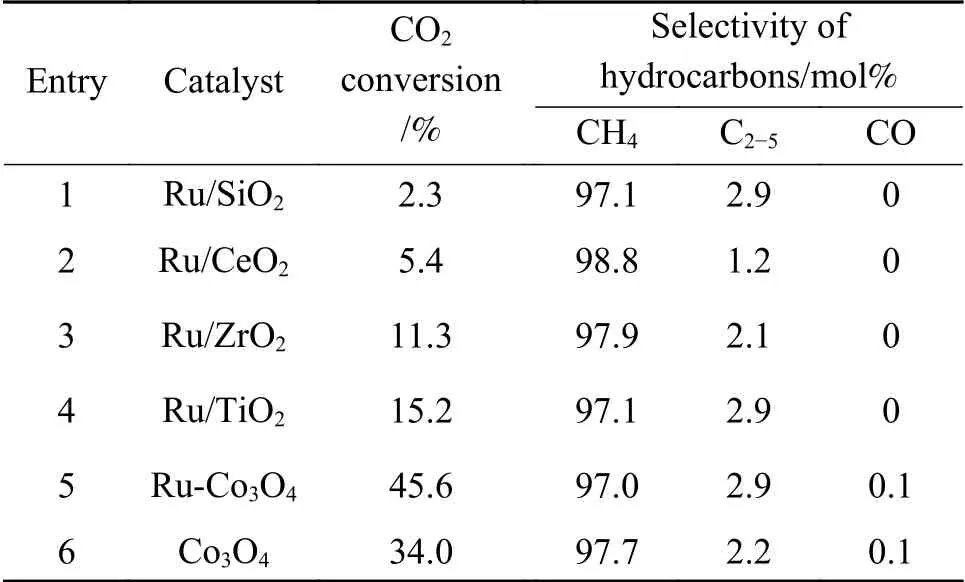
表1 不同Ru-基催化劑液相CO2 加氫制甲烷的催化性能Table 1 Catalytic results of various supported Ru catalysts for liquid-phase hydrogenation of CO2 to CH4
2.2 溶劑對Ru-Co3O4 反應性能的影響
鑒于Ru-Co3O4催化劑顯示出優良的催化性能,下面以Ru-Co3O4催化劑來研究不同溶劑對反應性能的影響規律。表2 為水、正丁醇、1,4-丁內酯、二甲基甲酰胺(DMF)、正壬烷、異辛烷和十氫萘溶劑中,Ru-Co3O4的催化反應結果。不同溶劑中Ru-Co3O4催化劑上的主要加氫產物均為CH4,其選擇性在87%-99%;除CH4外,還有少部分C2-5烴類產物的產生。
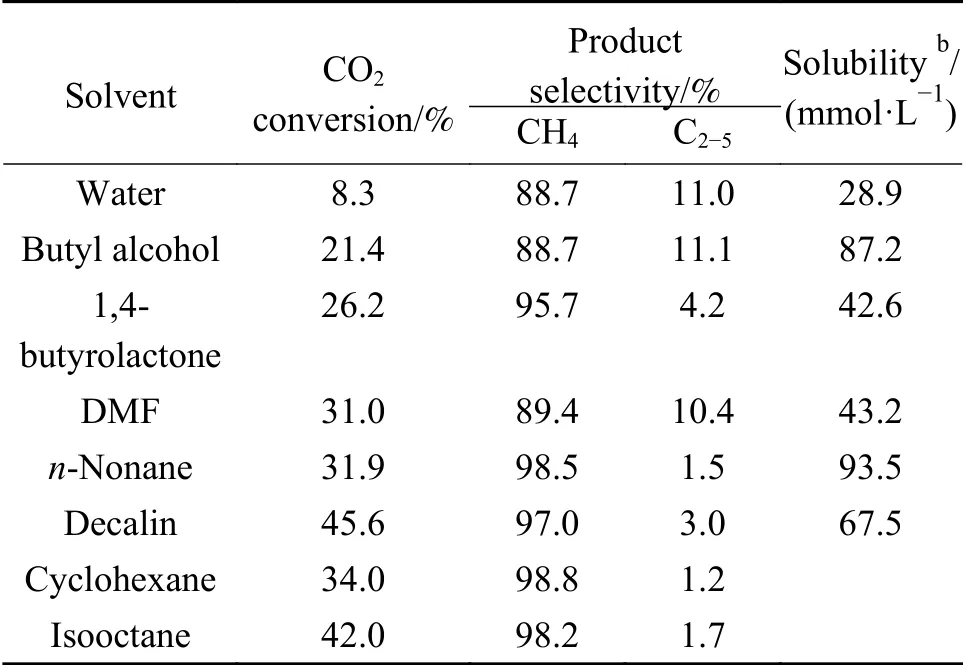
表2 不同溶劑中Ru-Co3O4 催化劑上CO2 液相加氫制甲烷性能的影響aTable 2 Catalytic results of Ru-Co3O4 for liquid-phase hydrogenation of CO2 to CH4 in different solventsa
由表2 可知,溶劑對Ru-Co3O4催化劑的活性具有顯著影響。水作為溶劑Ru-Co3O4表現出較低的活性,僅有8.3%;而在正丁醇中,其催化活性提高1.5 倍左右,CO2轉化率達到21% 左右;以1,4-丁內酯和DMF 為溶劑,其催化活性可進一步提高,CO2轉化率分別達到26%和31%左右。一般來說,多相催化劑的加氫反應活性常常依賴于其表面活性氫的濃度。催化劑表面活性氫濃度越高,反應速率越快,同時表現出較高的活性[20]。在氣相反應中,增大氫壓往往有利于提高催化劑表面的活性氫濃度,進而提高催化劑的加氫活性[21];與之類似的是,在液相反應中,溶劑溶解H2的能力越高,則越有利于提高催化劑表面活性氫的濃度。因此,作者推測,Ru-Co3O4催化劑在有機溶劑中的活性比在水中的高,主要是由于反應物H2分子在有機溶劑里具有更高的溶解度所致。
因此,考察了H2分子在上述溶劑中的溶解度。為了更好地模擬真實的反應條件,在溶解度實驗中,H2的壓力與反應時的壓力相同。由表2可知,在水中,H2的溶解度較低,為28.9 mmol/L;H2在正丁醇、1,4-丁內酯和DMF 等溶劑中的溶解度是水中的1.5-3.0 倍。這一結果初步證實了作者的推測,即有機溶劑高的溶氫能力提高了Ru-Co3O4的催化活性。為了進一步證實作者的猜測,選取正壬烷作為反應溶劑。發現H2在正壬烷中的溶解度也較高(表2)。催化評價結果顯示,Ru-Co3O4催化劑在正壬烷中確實也表現出了較高的活性,其CO2轉化率達到32%左右。當十氫萘為反應溶劑時,雖然H2在十氫萘中的溶解度低于在正壬烷中的溶解度,但催化活性卻提高了50%左右,CO2轉化率達到了46%左右(表2)。
2.3 溶劑作用機制
為了研究溶劑作用,選取正壬烷和十氫萘作為研究對象。以D 取代H,在200 °C 考察了溶劑的氘代率。十氫萘和正壬烷的GC-MS 分析(所用計算方法參見文獻[22])結果表明,十氫萘的氘代率為4.8%,而正壬烷的氘代率只有3%。十氫萘分子結構中同時含有成環的碳原子和叔碳原子,不同碳原子上的氫在核磁共振氫譜中的化學位移相近,不易區分。為了準確確定哪個位置的碳原子的氫更易氘代,選擇分別具有成環碳原子的環己烷和含有叔碳的異辛烷作為溶劑,并考察了它們的加氫反應性能。
由表2 可知,環己烷溶劑中CO2轉化率只有34%,而在異辛烷中CO2轉化率達到了42%。作者推測叔碳原子上的氫活潑性高,容易發生氘代。因此,叔碳原子可能是交換和傳遞活性氫的關鍵位點。在核磁共振氫譜中化學位移相差較大的異辛烷的氘代實驗證明了這一點。圖3 是氘代異辛烷和未氘代異辛烷的氫核磁共振譜定量分析。可以發現,氫在叔碳上的氘代率最高,達7.3%,仲碳上的氫平均氘代率次之,為6.0%,而伯碳上的氫平均氘代率僅有0.6%。這一結果與十氫萘的氘代實驗結果相吻合,說明叔碳是交換和傳遞活性氫的最活潑的位點。除叔碳外,仲碳也具有交換和傳遞活性氫的能力。
十氫萘具有較強的傳遞活性氫的能力(“傳遞氫”),那它是否可以將本身的氫作為活性氫進行加氫反應,即起到“供氫”的作用呢?這一作用要求十氫萘首先能夠將氫化學吸附到催化劑表面,并活化和傳遞氫。圖4 為十氫萘在不同樣品表面吸附的漫反射紅外光譜。在Ru-SiO2催化劑存在時,通入十氫萘后,譜圖在2858 和2929 cm-1出現了振動峰,分別對應十氫萘中的-C-H 和-CH2-伸縮振動征峰[23],Ar 吹掃后,這些特征峰消失,說明十氫萘在Ru-SiO2表面沒有發生化學吸附。然后,以相同的方式將十氫萘分別通入裝有Co3O4和Ru-Co3O4的原位池中進行飽和吸附,Ar 充分吹掃后譜圖在2851 和2922 cm-1仍然有十氫萘的-C-H和-CH2-的伸縮振動征峰;相較于Ru-SiO2上物理吸附的十氫萘,這些特征峰的峰值向低波數偏移大約7 cm-1,說明十氫萘化學吸附于Co3O4和Ru-Co3O4的表面。Co3O4上十氫萘和Ru-Co3O4催化劑上十氫萘的吸附峰位置相同,說明Ru 的引入對十氫萘的化學吸附沒有明顯的影響。
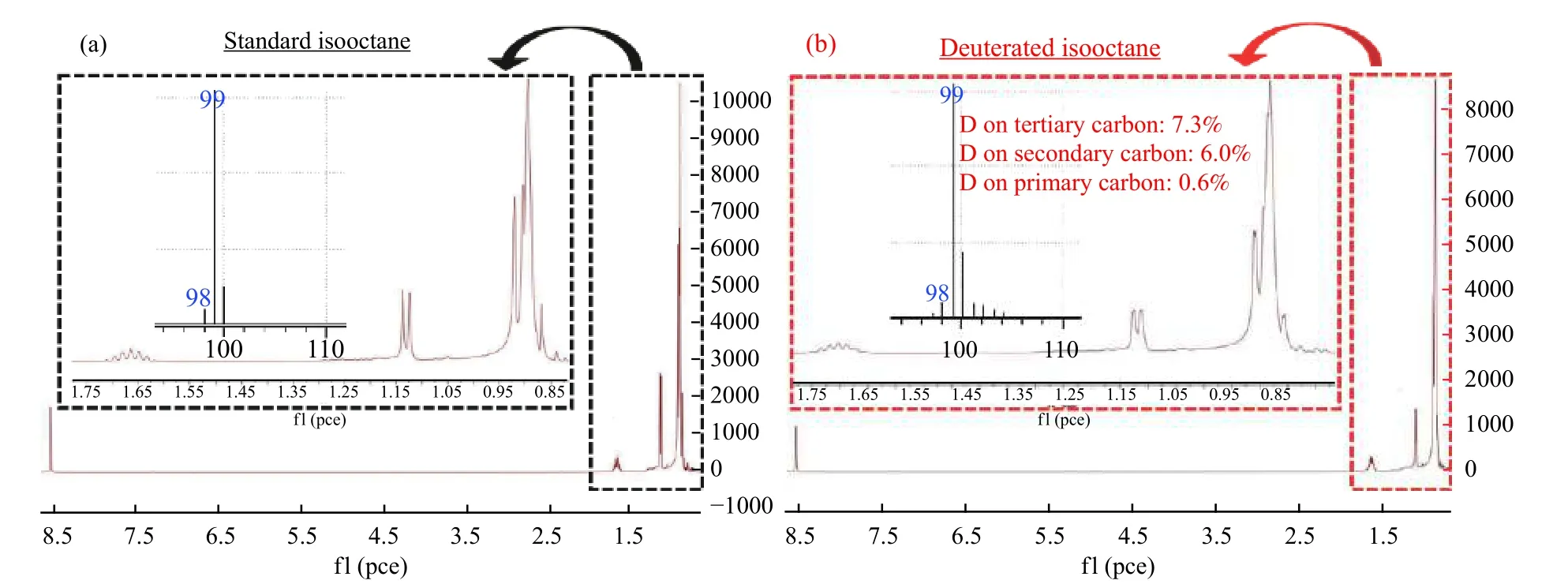
圖3 異辛烷標樣(a)和氘代異辛烷(b)的核磁共振色譜Figure 3 Nuclear magnetic resonance chromatograms of standard isooctane (a) and of deuterated isooctane (b)
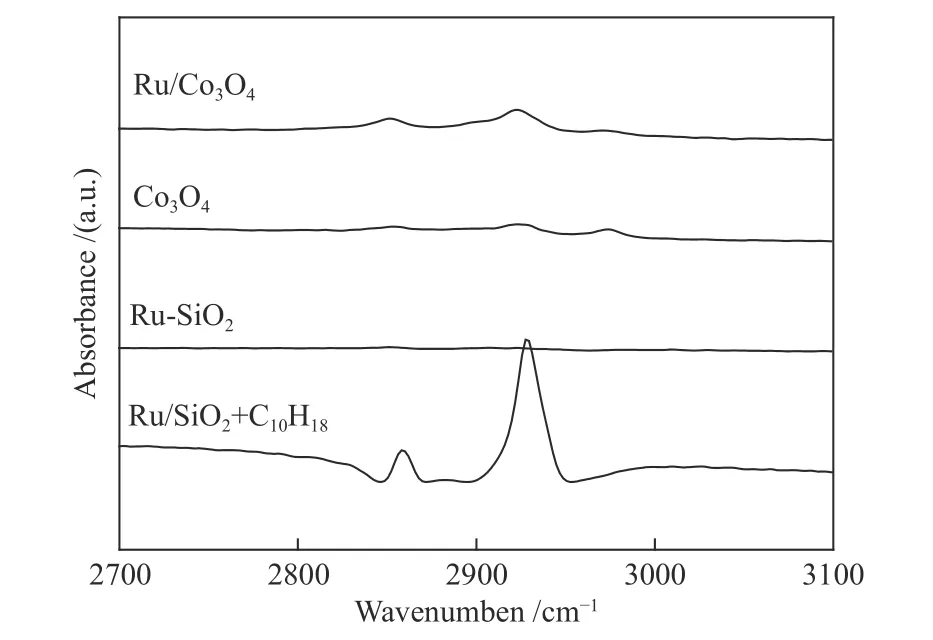
圖4 室溫下不同樣品吸附十氫萘的漫反射紅外吸附譜圖Figure 4 DRIFTS spectra for adsorption of decalin on the surfaces of various samples at room temperature
以原位漫反射紅外光譜研究了Ru-Co3O4催化劑上CO2加氫反應過程,見圖5。發現在200 °C時,將催化劑分別置于CO2和十氫萘的氛圍中,沒有CH4的特征峰。而將催化劑置于同時含有CO2和十氫萘的氛圍中,在3015 cm-1出現了甲烷峰(圖5(a)),這與作者測得的甲烷標準峰位置一致,同時在1000-1300 cm-1發現含碳物種的特征峰[23],這說明了CO2與十氫萘上的氫可以反應生成CH4,十氫萘具有“供氫”的作用。進而作者又考察了,Ru-Co3O4催化劑上CO2和十氫萘在不同溫度下的反應行為。如圖5(b)所示,室溫下沒有任何紅外峰,說明室溫下沒有反應發生;隨著反應溫度升高至90 °C,在1000-1300 cm-1出現紅外振動峰,可以歸屬于吸附于催化劑表面的含碳物種。當溫度進一步升高至160-200 °C 時,在3015 cm-1處出現了CH4特征峰,說明CO2加氫生成CH4需要在較高溫度下才能進行。值得注意的是,在200 °C反應30 min 之后,氣相甲烷的特征峰消失,同時十氫萘的特征峰也逐漸減弱(2800-2900 cm-1)。在30 min 后,氣相和物理吸附的峰消失。而離子態的十氫萘的特征峰仍存在,但十氫萘離子中沒有活性氫,不能繼續反應生成CH4。
為了進一步研究反應機理,對催化劑進行了H2-TPR 表 征。由 圖6 可 知,Co3O4分 別 在242 和390 °C 存在兩個還原峰,第一個歸屬于Co3O4還原為CoO 的峰;第二個歸屬于CoO 還原為Co 的峰[24]。Ru 的引入使Co3O4的還原溫度向低溫方向發生了明顯的偏移,這是由于Ru 具有良好的活化氫氣的能力,產生的活性氫通過氫溢流促進了Co3O4的還原。
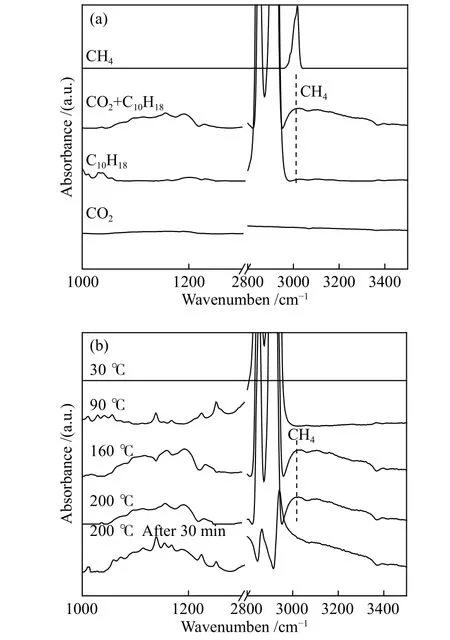
圖5 Ru-Co3O4 催化劑表面CO2 加氫的漫反射紅外譜圖:(a) 200 °C; (b) 不同溫度下Figure 5 DRIFTS spectra of CO2 hydrogenation to CH4 at 200 oC on Ru-Co3O4 in CO2, decalin, (CO2+decalin), and DRIFTS spectrum of standard CH4 (from the bottom to the top direction) (a); and DRIFTS spectra of CO2 hydrogenation to CH4 at 200 oC on Ru-Co3O4 in (CO2+decalin) at different reaction temperatures (b)
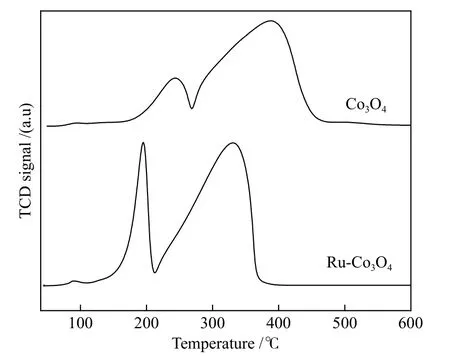
圖6 催化劑的H2-TPR 譜圖Figure 6 H2-TPR profiles of catalyst
以上結果表明,十氫萘起到活性氫的載體作用,即起到“供氫”的作用。H2的活化主要有兩條路線,一條是氫氣吸附在Ru 上活化生成活性氫并與CO2反應生成甲烷(圖7(a));另一條是H2被吸附在Co3O4表面的十氫萘離子(失去活性氫之后所形成的)活化,隨后與CO2反應生成CH4(圖7(b))。實際上,十氫萘作為“供氫”溶劑已有報道,認為十氫萘的這種供氫作用是因為碳氫鍵的鍵能低于氫氫鍵的鍵能[25]。另外,Ru-Co3O4催化劑在300 °C還原后形成少量平均粒徑約23 nm 的金屬Co 顆粒,也會對H2的活化起到一定的作用。
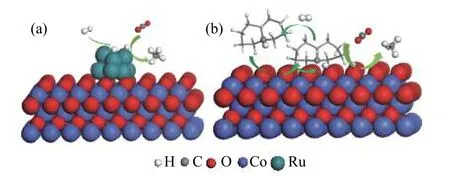
圖7 十氫萘溶劑中Ru-Co3O4 表面CO2 加氫生成甲烷的反應路徑Figure 7 Schematic presentation for catalytic mechanism of CO2 hydrogenation to CH4 over Ru-Co3O4 in decalin solvent
3 結 論
采用共沉淀法制備的Ru-Co3O4催化劑,在液相中CO2加氫制甲烷反應中顯示出優良的催化性能。與傳統浸漬法負載的Ru 基催化劑相比,其具有高的加氫活性和甲烷選擇性,以十氫萘為溶劑,在200 °C 及H2/CO2= 3∶1(v/v, 4 MPa)條 件 下,其CO2轉化率達到45.6%,CH4的選擇性達到97%左右。同位素標定實驗和原位紅外光譜表明,十氫萘和異辛烷對催化活性具有明顯的促進作用。十氫萘首先起到交換和傳遞活性氫的作用,即起到“活化氫”的作用,從而有利于提高反應的活性;相比于伯碳和仲碳原子,叔碳原子上的氫活性更高,說明叔碳是交換和傳遞活性氫的最活潑的位點。H2在Ru-Co3O4催化劑表面活化主要有兩條路線,一條是氫氣吸附在Ru 上活化生成活性氫并與CO2反應生成甲烷;另一條是H2被吸附在Co3O4表面的十氫萘離子活化,再與CO2反應生成CH4,兩條路徑同時作用使反應活性大幅提高。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
