SOD1G72S基因突變致散發型肌萎縮側索硬化癥1例
2021-01-26 06:26:48趙京燕李建軍張強勝潘朝
疑難病雜志 2021年1期
關鍵詞:基因突變
趙京燕,李建軍,張強勝,潘朝
作者單位: 050091 石家莊,河北以嶺醫院肌萎縮專科
患者,女,64歲。因進行性肢體無力7月余于2020年4月2日入院。患者于2019年9月勞累后出現左腳背屈力弱,并逐漸出現右下肢無力。當地醫院查自身免疫性周圍神經抗體均為陰性,腦脊液細胞分析未見異常,腦脊液蛋白68.0 mg/dl,IgG 47.6 mg/L,血IgE 273.0 IU/ml;頭顱MR示多發缺血灶;肌電圖示左下肢神經運動傳導波幅減低,潛伏期及速度正常,左脛前肌、腓腸肌、股四頭肌,右腓腸肌均呈神經源性損害(靜息狀態下可見大量纖顫正銳波,小力狀態下可見巨大電位)。給予甲潑尼龍80 mg靜脈滴注并逐漸減量,癥狀無改善。2019年12月于外院診斷“可疑運動神經元病”,給予丙種球蛋白靜脈滴注5 d,效果不佳。后患者雙下肢逐漸癱瘓不能活動,腰部無力不能端坐。2020年1月累及雙上肢,咳嗽力減弱,底氣不足,自覺腹部有束帶感,無明顯胸悶氣短,為明確診斷遂收入院。否認家族遺傳病史及類似疾病史。入院后查體:雙下肢肌肉萎縮松弛明顯,雙上肢肌力4級,雙側肱二頭肌、肱三頭肌腱反射(+),雙下肢肌力1級,雙足背屈力0級,雙下肢肌張力減低,雙側膝腱反射、Hoffmann征、掌頜反射、Babinski征均(-),深淺感覺未見明顯異常。復查肌電圖:左正中神經、左尺神經MCV波幅較對側減低,左腓總神經MCV未引出,右腓總神經MCV波幅減低,雙脛神經MCV減慢,波幅減低,雙腓總神經、雙脛神經F波未引出,廣泛性神經源性異常。
采用NGS+PCR-Sanger測序驗證技術對肌萎縮側索硬化65個相關基因的外顯子編碼區進行檢測,顯示SOD1基因外顯子3存在 c.217G>A(p.Gly72Ser)雜合變異(圖1),確診為“散發性肌萎縮側索硬化癥(SALS)”,患者子女未進行相關檢測。治療給予利魯唑 50 mg口服,每天2次,以延緩疾病發展。2020年6月24日回訪,患者雙上肢已癱瘓不能活動,并出現明顯的氣短,不能平躺,指尖血氧飽和度91%,在服用利魯唑基礎上配合間斷無創呼吸機輔助呼吸。
討 論肌萎縮側索硬化癥(amyotrophic lateral sclerosis,ALS)是一種原因未明且罕見的神經系統變型致死性疾病,90%以上為散發型病例(sporadic ALS,SALS),遺傳型或家族性(family ALS,FALS)占5%~10%,以進行性加重的骨骼肌無力、肌萎縮、肌束顫動、延髓麻痹和錐體束征為主要臨床表現,最終多死于呼吸肌麻痹或并發的呼吸道感染,平均病程3~5年。ALS的發病機制復雜,涉及到多種病理生理機制,如遺傳機制、興奮性氨基酸毒性、氧化應激損傷、軸突運輸障礙、線粒體功能障礙、蛋白質異常聚集、膠質細胞炎性反應等。國內外學者普遍認為ALS的發病是基因與環境共同作用的結果,目前已發現ALS相關的致病基因有33種,其中定位于21號染色長臂的SOD1基因突變率為2%~7%,約 20%的FALS和 5%的SALS與該基因突變有關[1]。2015年再修訂的El Escorial診斷標準中明確提出,如果基因檢測發現已知的ALS相關基因的致病突變,即可確診為ALS。
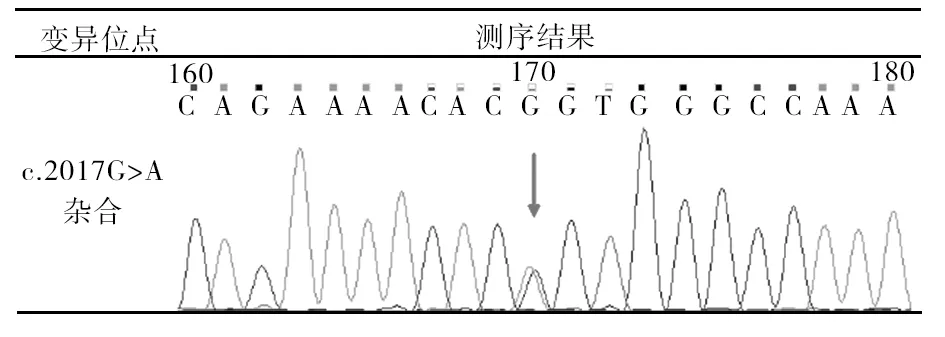
圖1 患者基因測序結果
本患者病情進展迅速,發病9個月時四肢癱瘓并出現呼吸困難,利用高通量測序技術對ALS相關基因的外顯子編碼區進行檢測,結果提示SOD1 基因外顯子3存在c.217G>A(p.Gly72Ser)雜合變異,為致病突變,故確診為SALS,并給予利魯唑口服。利魯唑是目前唯一證實可以在一定程度上延緩病情進展的藥物。
迄今國際上已報道150余種SOD1基因突變類型,國內僅報道了10余種 SOD1 基因突變[2-7],如Cys111Ty、Cys146X、Gly147Asp、p.IIe150Thr、p.Ala4Val、H46R、E133V、V29A、N86I等,主要分布在2、4和5號外顯子,其中1例病情進展快速的散發型ALS患者發現位于3號外顯子的雜合G72C突變,與本例的雜合G72S突變是相同位置其他點變異。國外關于SOD1G72S基因突變在肌萎縮側索硬化癥在線數據庫(ALSoD)記錄有2例,其中1例是在FALS中檢出,而另外1 例在 SALS中檢出,且發病15個月時死于呼吸衰竭[8-9]。
總之, 本例散發型肌萎縮側索硬化癥患者檢出致病基因突變SOD1G72S,既往國內文獻尚未見報道,對研究中國 ALS 患者 SOD1 基因突變特點和分布規律有一定幫助。
猜你喜歡
英語世界(2023年6期)2023-06-30 06:29:10
中國醫學影像學雜志(2021年6期)2021-08-13 08:43:36
中國生殖健康(2020年2期)2021-01-18 02:51:26
小學生導刊(2018年13期)2018-06-29 03:49:00
中國生殖健康(2018年2期)2018-01-12 13:57:51
現代檢驗醫學雜志(2016年4期)2016-11-15 02:01:14
中國現代醫學雜志(2015年26期)2015-12-23 11:04:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:44
中華皮膚科雜志(2014年4期)2014-12-19 12:55:49
中國神經精神疾病雜志(2014年1期)2014-03-01 03:23:22
