蒽熱生焦初期的反應分子動力學模擬
2021-01-05 07:04:34楊海洋范啟明王麗新
石油學報(石油加工) 2020年5期
關鍵詞:模型
楊海洋, 任 強, 范啟明, 周 涵, 王麗新
(中國石化 石油化工科學研究院,北京 100083)
針狀焦具有機械強度高、熱膨脹系數小、電阻率低、耐腐蝕等優點,主要用于高功率、超高功率石墨電極和負極材料的制造。因而,針狀焦是一種高附加值產品,其市場價格相比普通焦高得多。
針狀焦是大分子稠環芳烴(PAHs)的類石墨晶體[1],微晶以層狀堆積,單元取向度高。按照原料來源,針狀焦可分為石油系針狀焦和煤系針狀焦。1950年美國大湖炭素公司首先發明了石油系針狀焦[2],開啟了針狀焦的應用與研究。1965年Brooks等提出的炭質中間相理論[3-4]為炭素科學提供了理論研究基礎,也推動了針狀焦生產技術的發展。中國對針狀焦的研究始于20世紀70年代末,中國石化石油化工科學研究院先后完成了石油系針狀焦制備的小試、中試和工業試驗,確定原料和焦化工藝,建立了相應的測試方法[5-6]。
目前,中國針狀焦生產還存在質量不穩定、焦炭強度低、粉焦多等問題,與國際先進水平仍有差距[7]。為了提升針狀焦品質,需要從針狀焦生成機理著手研究,考察針狀焦生成的反應過程,明確對提升針狀焦品質有利的影響因素。針狀焦生成過程主要遵循液相炭化機理,在700~780 K的高溫下,相對分子質量較小的芳香烴(如:萘、蒽、菲、芘)經過脫氫縮合逐漸生成大平面稠環芳烴[8-14]。
為深入研究針狀焦的生成機理,選擇蒽作為模型化合物,研究蒽熱生焦過程的反應分子動力學。在液相炭化的反應溫度下,蒽分子很難直接脫氫進而均裂生成自由基,比較容易發生氫轉移反應,進而引發自由基反應[15]。為考察蒽熱生焦過程的反應分子動力學和熱力學,采用分子模擬方法研究蒽液相炭化生成四聚物的反應歷程。
1 計算模擬方法
針狀焦生成過程是一個復雜的物理化學過程,反應階段主要是原料受熱后逐漸縮合生成低聚物。因此,需要分階段進行建模和模擬,本研究主要剖析蒽生成二聚物、二聚物生成三聚物和二聚物生成四聚物等3個階段的蒽熱生焦反應過程。
1.1 創建模型
采用Materials Studio 2017 R2軟件(簡稱MS)創建蒽分子的模型。首先選用MS中的Amorphous Cell模塊創建一個包含200個蒽分子的三維周期性初始模型,其密度初步設定為0.001 g/cm3;然后選用Forcite模塊對初始模型進行結構弛豫,力場選用Compass II,精度為Medium。先選擇正則系綜(NVT)在2000 K下模擬分子動力學200 ps,促使蒽分子充分散開,在1500~2500 K溫度區間選擇NVT系綜模擬退火(Annealing);然后選擇等溫等壓系綜(NPT)在770 K和1 GPa下模擬分子動力學200 ps,在500~1000 K溫度區間選擇NPT系綜進行退火模擬;再選擇NPT系綜在770 K和0.2 MPa下模擬分子動力學200 ps,選擇NVT系綜在770 K和0.2 MPa下模擬分子動力學200 ps;最終得到密度為0.799 g/cm3的純蒽模型。采用相同方法創建含有100個蒽與50個蒽二聚物的混合體系模型和含有100個蒽二聚物的模型,結構弛豫后的密度分別為0.846 g/cm3和0.948 g/cm3。3種模型如圖1所示。
1.2 反應分子動力學模擬
將模型從MS軟件中導出,導入MAPS軟件(Materials and Processes Simulations 4.1,SCIENOMICS公司產品)中。選用MAPS軟件的Lammps[16]模塊,采用反應分子動力學方法ReaxFF[17]模擬蒽熱生焦過程中的反應分子動力學,力場參數選自含有C、H元素的反應力場文件CH_aromatics.ff[18-20]。針狀焦生產溫度一般在700~780 K[21-27]。根據溫度加速反應動力學理論[28]將模擬溫度提升至2600 K[29]。反應分子動力學模擬選擇正則系綜(NVT),步長0.1 fs,運行時長200 ps,每1 ps取1幀畫面。使用MAPS中ReaxFF分析插件對反應分子動力學模擬結果進行分析。
1.3 量子化學計算分析
由于采用升溫加速動力學的方法,模擬中可能會發生低溫下不會發生的反應而出現失真現象。因此,有必要采用量子化學方法對某些關鍵反應進行計算分析。量子化學方法的計算分析選用MS軟件中Dmol3模塊,通過Dmol3模塊可以優化模型分子的幾何結構,搜索基元反應的過渡態,計算反應能壘和反應焓變。計算基于密度泛函理論(Density Function Theory)的量子化學方法進行,具體方法選用基于廣義梯度近似(GGA)的PBE (Perdew-Burke-Ernzerhof)泛函,在大數值基組DNP(雙數值軌道基組+p軌道極化函數)水平上選擇全電子(All electron)進行計算。自洽場(Self-Consistent Field, SCF)迭代收斂閾值設為1×10-5Ha(1 Ha=2625.5 kJ/mol),軌道截斷半徑為0.37 nm,收斂精度設為:能量2×10-5Ha,受力0.04 Ha/nm,位移5×10-4nm。過渡態搜索采用完全線性同步和二次同步變化方法(Complete LSF/QSF)。
2 結果與討論
2.1 純蒽模型反應分子動力學模擬
2.1.1 純蒽模型反應分子動力學
圖2為純蒽模型反應分子動力學模擬結果。其中,圖2(a)和(b)分別為模擬100 ps和200 ps時模型的結構狀態;圖2(c)和(d)分別為將圖2(a)和(b)中未反應的蒽分子隱藏后,模擬100 ps和200 ps時生成的分子和自由基的結構狀態。不同顏色對應不同種類的分子或自由基,顏色種類越多說明生成的分子和自由基種類越多。由圖2純蒽模型反應分子動力學模擬結果可知:純蒽熱生焦過程發生了縮合、氫轉移和重排等反應,很少發生開環反應;100 ps時反應生成了·C28H19、·C14H9、·C14H11等自由基;200 ps時反應生成了·C28H20、·C28H19、·C14H9、·C14H11、C28H18等自由基或分子,·C28H19、·C14H11數量增多。
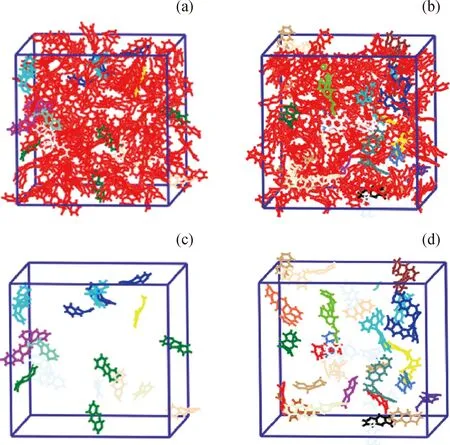
圖2 蒽反應分子動力學模擬結果Fig.2 Reactive molecular dynamics simulationof anthracene coking process(a) Structure diagram at 100 ps; (b) Structure diagram at 200 ps;(c) Molecules and free radicals generated in 100 ps;(d) Molecules and free radicals generated in 200 psVarious colors stand for different molecular or radical types
使用MAPS中ReaxFF分析插件對蒽熱生焦反應分子動力學模擬結果進行分析。蒽熱生焦反應模擬中蒽分子數量和生成物(·C28H20、·C14H9、·C14H11、H2、C28H18等)數量隨模擬時間的變化曲線如圖3所示。由圖3可觀察到,蒽分子數量隨著模擬時間的增長而逐漸減少,從200個減少到160個左右,總共消耗了大約40個蒽分子,模擬200 ps時轉化率約為20%。由圖3還可以觀察到:·C28H20數量最多時只有3個,約占原料分子數量的1.5%,對后續反應影響很小;·C14H9、·C14H11和·C28H19數量最多時分別有7、10和5個,數量較多,說明這些自由基穩定性較強,對后續反應影響較大;反應生成了1個C28H18分子,為蒽分子脫去2個氫原子后生成的二聚物。
根據模擬所取的幀數統計反應次數,幀數越多,統計到的反應次數可能會越多。模擬中每1 ps取一幀,模擬時長200 ps共取得200幀。其中,發生次數較多的反應如表1所示。由表1可知:蒽熱生焦模擬過程中反應次數最多的反應是2個蒽分子的縮合反應(A1),其生成的·C28H20自由基熱穩定性差,通過逆反應(A2)快速生成C14H10;其次是2個蒽分子之間發生氫轉移反應(A3),生成了·C14H9和·C14H11,其穩定性都較強;蒽分子和自由基 ·C14H9結合生成·C28H19的反應(A4)次數也較多,生成·C28H19。
2.1.2 量子化學分析
蒽分子參與反應的部位主要是1號位、2號位和9號位的碳,很少發生在11號位[30-31]。根據量子化學第一性原理計算蒽分子結構性質和凈電荷分布,結果如圖4所示。由圖4可知:蒽分子中1號位、2號位和9號位C-H鍵的鍵長相差不大,鍵長無法體現1號位、2號位和9號位上C-H鍵強度的差異;而9號位的C比1號位和2號位的C上分布的凈電荷稍多,表明9號位的C活性稍強;3個位置上的H電荷分布相當,表明3個H原子的活性相同。Sasaki等[30-31]計算蒽分子中1號位、2號位和9號位C上的自由價分別為0.459、0.408、0.520,認為蒽分子9號位活性最強。綜合分析,9號位C活性最強,易發生脫氫反應或者氫轉移反應生成自由基,與文獻[32]結論一致。

圖3 蒽熱生焦反應中反應物和生成物數量隨模擬時間的變化曲線Fig.3 Numbers of anthracene molecules and ·C28H20, ·C14H9, ·C14H11, ·C28H19, C28H18 versus simulation time(a) Anthracene; (b) ·C28H20, ·C14H9, ·C14H11, ·C28H19 and C28H18
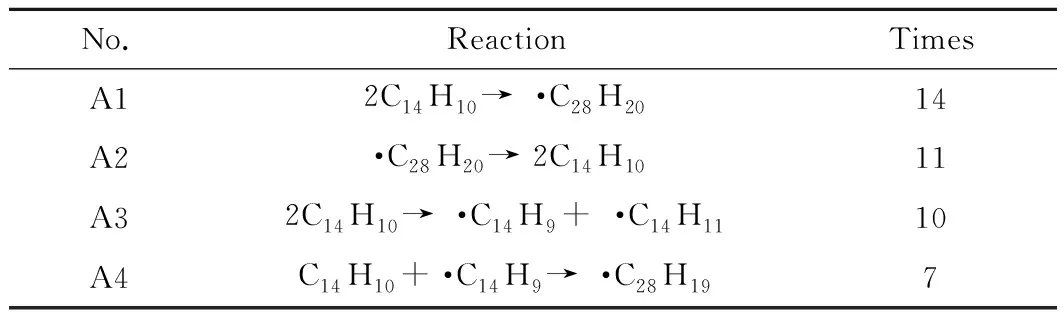
表1 模擬過程中部分反應統計Table 1 Statistics of partial reactions during the simulation
通過量子化學計算可知:1號位、2號位和9號位C-H鍵的均裂能分別為483.25、484.70、483.60 kJ/mol,均裂能都很高。這說明3個位置上C-H鍵斷裂的難度相當,都需要很高的溫度。依據Poutsma規則[33-34],芳烴很難在780 K以下均裂生成自由基,一般通過氫轉移反應引發生成自由基。
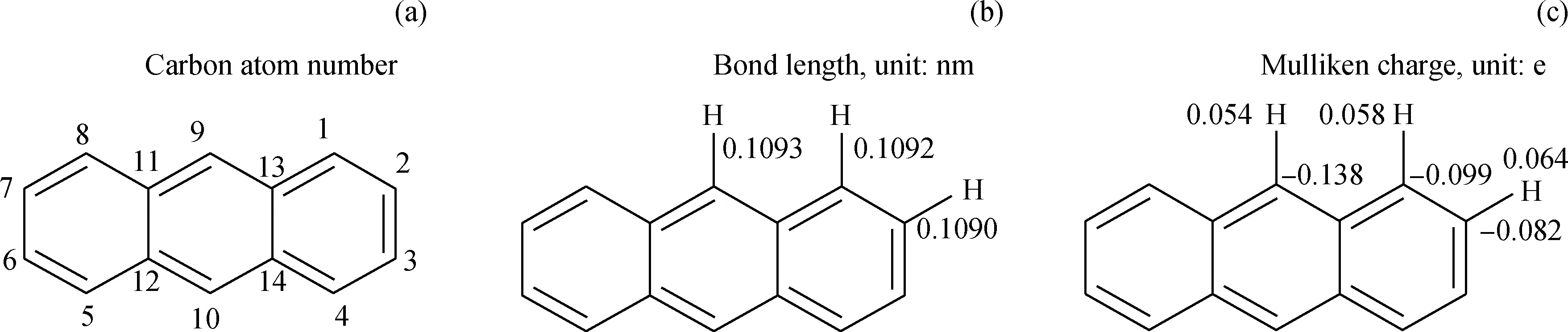
圖4 蒽分子結構性質和電荷分布Fig.4 Anthracene structure features and Mülliken charge(a) Carbon atom number; (b) Bond length; (c) Mülliken charge of atom
蒽受熱以后可能發生多種反應[25]。為避免采用升溫加速動力學方法模擬蒽熱生焦反應可能導致的模擬失真問題,采用量子化學對反應分子動力學模擬過程中發生的各種反應進行動力學和熱力學分析,通過搜索過渡態方法計算反應的能壘和焓變,結果如表2所示。
由表2可知,2個蒽分子之間首先發生氫轉移反應(B1),引發生成自由基。其反應能壘較高,為321.81 kJ/mol,反應焓變296.89 kJ/mol,為吸熱反應;因此該反應在溫度較高時才能夠發生。蒽分子間氫轉移反應能壘是蒽熱生焦反應過程中的第一道反應能壘,對后續反應影響較大。氫轉移反應可引發自由基,因而適當的供氫能夠穩定自由基的反應性,使反應中間體保持良好的流動性和溶解性,促使片層分子有序排列[35]。
蒽分子與自由基·C14H9結合生成·C28H19(B2),反應能壘很低,為10.81 kJ/mol,反應焓變為-165.29 kJ/mol,因而是易于發生的放熱反應,且對蒽二聚物的生成有較大影響。生成的·C28H19可能與周圍的蒽分子碰撞而失去1個氫原子,生成熱力學性質更為穩定的二聚物C28H18(B3)。其反應能壘為207.99 kJ/mol,反應焓變分別為17.29 kJ/mol,反應能壘較高,但在受熱時可以發生。
2個蒽分子縮合生成自由基·C28H20的反應(B4)為可逆反應,其正反應的能壘為197.47 kJ/mol,焓變為194.99 kJ/mol。根據微觀可逆性原理,正反應和逆反應經過同一活化體,因此該反應逆反應的能壘為2.46 kJ/mol,焓變為-194.99 kJ/mol。該逆反應能壘非常低,且為放熱反應,易于發生。純蒽模型反應分子動力學模擬中正、逆反應次數相近,因而檢測到的·C28H20數量非常少。這也說明·C28H20穩定性很差,很容易發生逆反應生成蒽。此外,·C28H20也可能發生分子內氫轉移反應(B5),反應能壘較低,為116.50 kJ/mol;反應焓變為-21.59 kJ/mol,為放熱反應,因而也較容易進行。
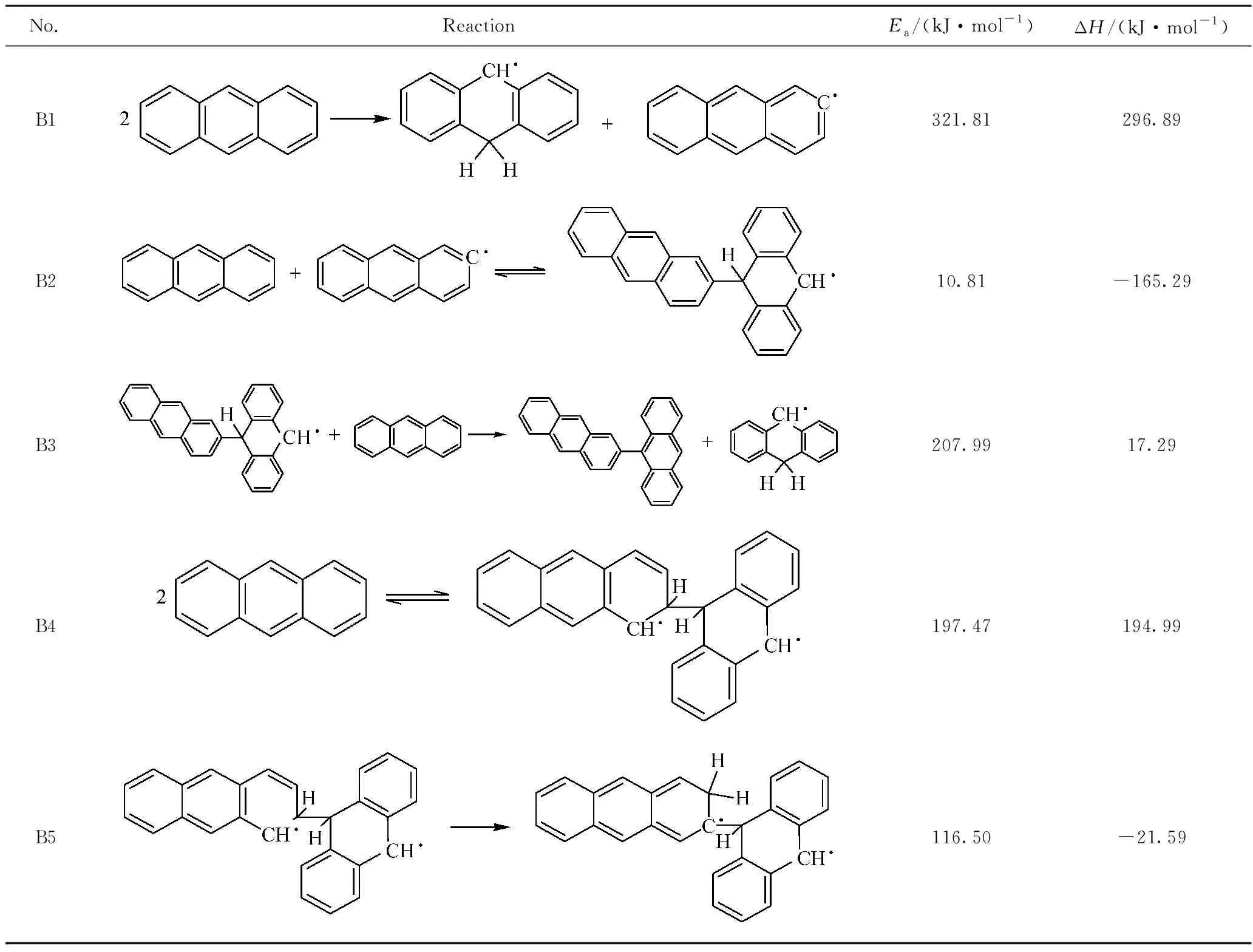
表2 蒽熱生焦反應能量分析Table 2 Energy analysis of anthracene pyrolysis
2.1.3 蒽縮合生成二聚物的反應歷程
根據純蒽模型反應分子動力學模擬和量子化學分析結果,可以推斷蒽熱生焦過程中生成二聚物的主要反應歷程如圖5所示,圖中數據為反應能壘。蒽先通過分子間氫轉移反應引發生成自由基·C14H9;·C14H9與蒽分子結合生成·C28H19;·C28H19被周圍的蒽分子奪去1個氫原子生成穩定的二聚物C28H18。反應過程中,自由基引發階段分子間的氫轉移反應能壘最高,是蒽熱生焦過程初期的控速步驟;相比自由基引發反應,自由基鏈傳遞反應能壘較低,較容易進行。
2.2 混合模型反應分子動力學模擬
圖6為混合模型反應分子動力學模擬結果。其中,圖6(a)為模擬100 ps時生成的分子和自由基的結構狀態;圖6(b)為模擬200 ps時生成的分子和自由基的結構狀態。圖中不同顏色對應不同種類的分子或自由基;顏色種類越多說明生成的分子和自由基種類越多。由圖6可知:混合模型模擬過程中發生了縮合、氫轉移、重排和開環等反應;100 ps時生成了·C28H17、·C28H19、·C28H20、·C14H9、·C14H11、·C42H27、C42H26等自由基或分子;200 ps時生成了·C28H17、·C28H19、·C14H9、·hC14H11、·C42H28等自由基,·C28H17、·C14H11數量有所增多。
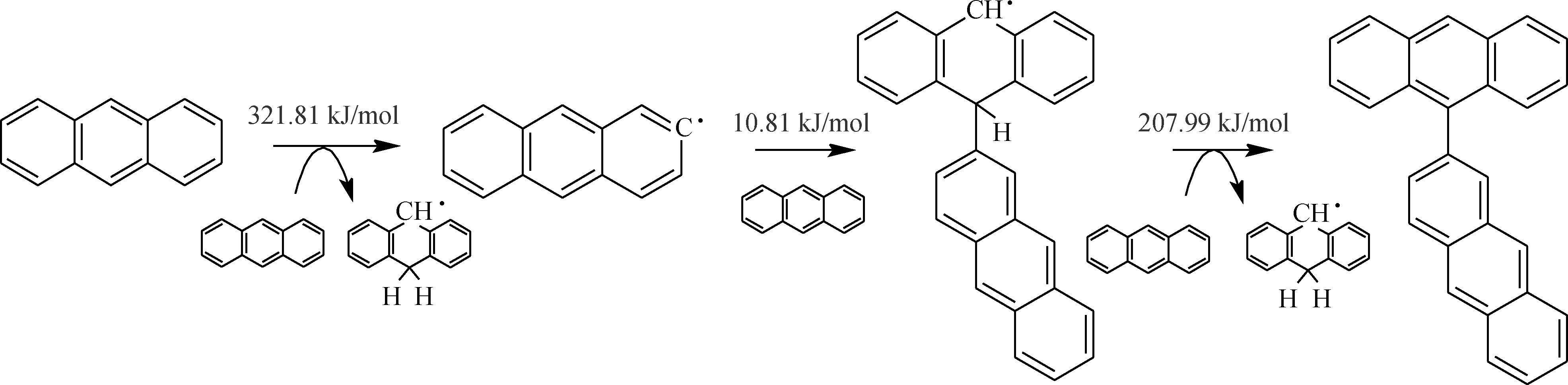
圖5 蒽縮合生成二聚物的反應歷程Fig.5 Reactive process of anthracene to dimerThe data in the diagram are reactive energy barriers.
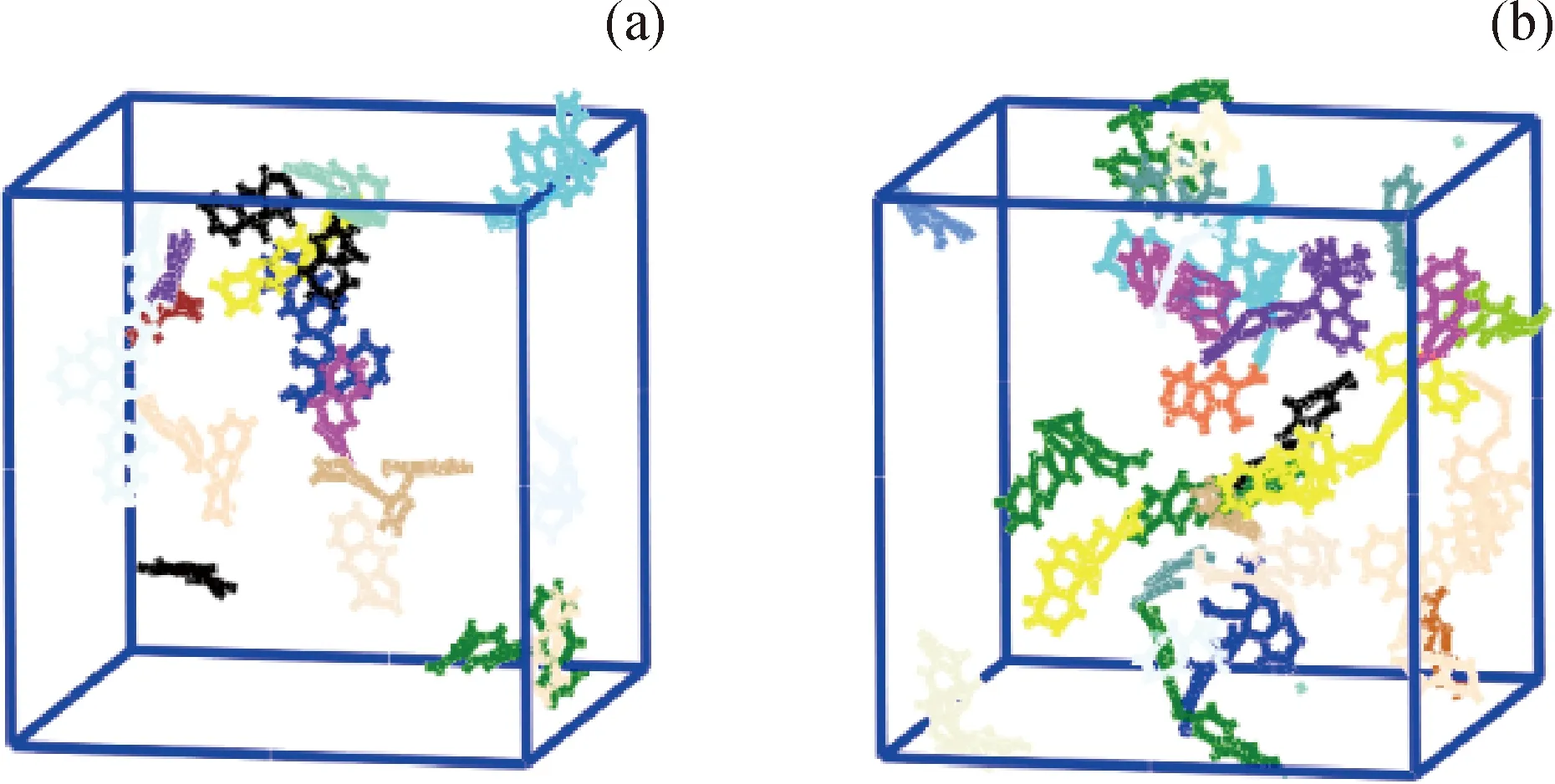
圖6 混合模型反應分子動力學模擬結果Fig.6 Reactive molecular dynamics simulation ofmix model coking process(a) 100 ps; (b) 200 psVarious colors stand for different molecular or radical types.
模擬中每1 ps取1幀,模擬時長200 ps共取得200幀。使用MAPS中ReaxFF分析插件對反應分子動力學模擬結果進行分析,其中部分反應次數統計如表3所示。蒽和二聚物分子數量隨模擬時間的變化曲線如圖7所示。
由表3可知:反應次數較多的反應是蒽分子的縮合反應及其逆反應(R1~R6);縮合生成的·C42H28、·C28H20、·C56H36自由基熱穩定性差,很容易發生逆反應;正、逆反應次數相當,累積生成的縮合產物分子數量很少,對后續反應影響很小。對后續反應影響較大的主要是分子間氫轉移反應(R7和R9)。氫轉移引發生成的自由基·C28H17和 ·C14H9可能與周圍蒽分子結合生成·C42H27(R8)和·C28H19(R10),也可能與周圍的二聚物分子反應生成三聚物C42H26(R11)。
由圖7可觀察到,蒽分子數量隨著模擬時間的增長而逐漸減少,從100個減少到85個左右,總共消耗了大約15個分子,模擬200 ps時轉化率約為15%。蒽二聚物分子數量從50個減少到40個左右,總共消耗了大約10個分子,模擬200 ps時轉化率約為20%。
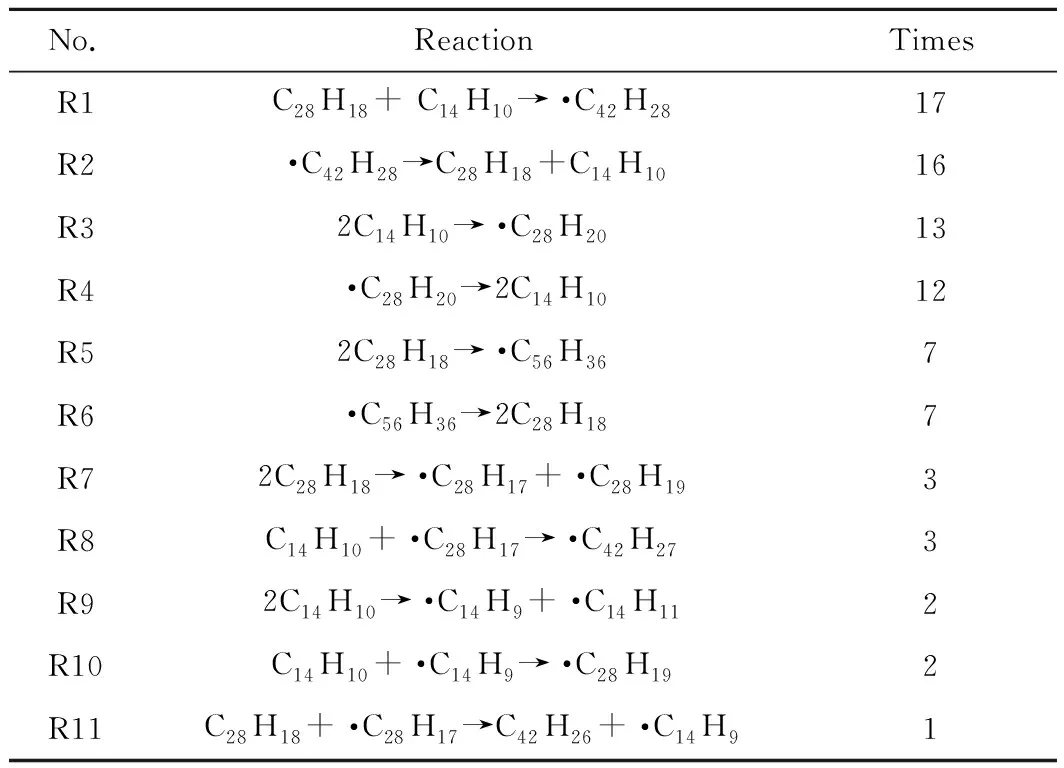
表3 混合模型模擬過程中部分反應統計Table 3 Statistics of partial reactions during the simulation
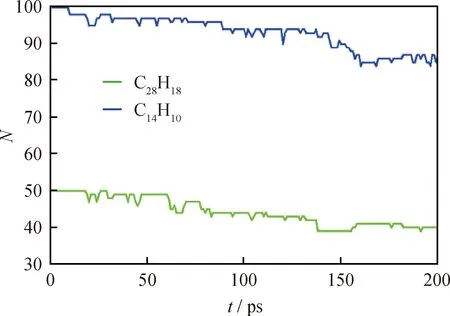
圖7 蒽和二聚物分子數量隨模擬時間的變化曲線Fig.7 Molecular numbers of anthracene anddimer versus simulation time
根據混合模型反應分子動力學結果可以觀察到蒽二聚物縮合反應的反應路線,如圖8所示。由圖8 可知,蒽二聚物先通過氫轉移反應引發生成自由基·C28H17,然后自由基·C28H17與周圍的二聚物分子反應生成三聚物。
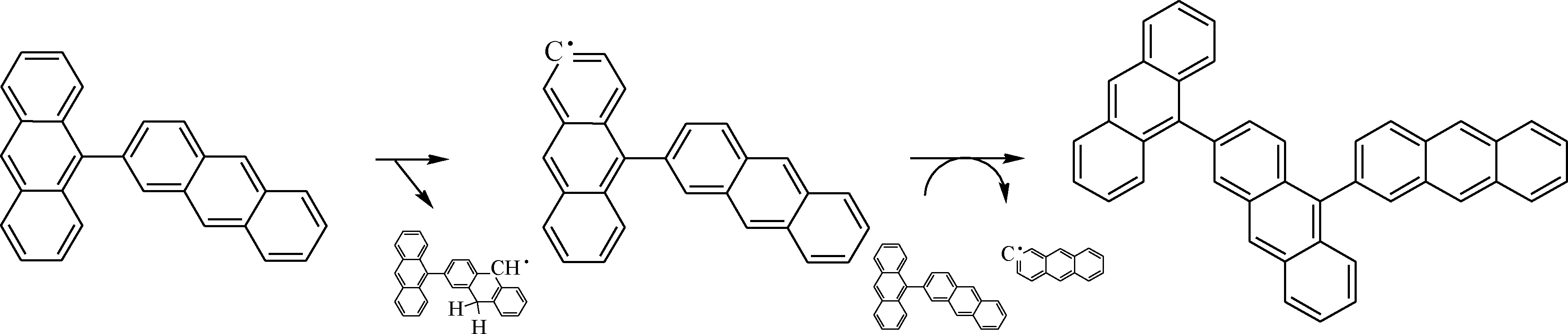
圖8 混合模型反應路線Fig.8 Reactive route of mix model in reactive molecular dynamics simulation
2.3 蒽二聚物模型反應分子動力學模擬
圖9為蒽二聚物模型反應分子動力學模擬結果。其中,圖9(a)為模擬100 ps時生成的分子和自由基的結構狀態;圖9(b)為模擬200 ps時生成的分子和自由基的結構狀態。圖中不同顏色對應不同種類的分子或自由基;顏色種類越多說明生成的分子和自由基種類越多。由圖9可以觀察到:模擬過程中發生了縮合、氫轉移、重排和開環等反應;100 ps時生成了·C28H17、·C28H19等自由基;200 ps時生成了·C28H17、·C28H19、·C56H35、·C56H36等自由基。
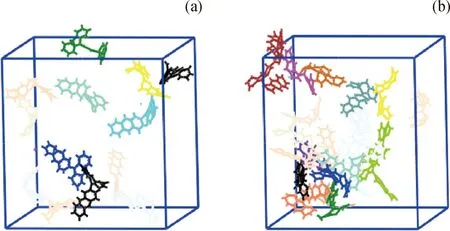
圖9 蒽二聚物模型反應分子動力學模擬結果Fig.9 Reactive molecular dynamics simulation ofanthracene dimer coking process.(a) Molecules and free radicals generated in 100 ps;(b) Molecules and free radicals generated in 200 psVarious colors stand for different molecular or radical types.
使用MAPS中ReaxFF分析插件對反應分子動力學模擬結果進行分析。模擬中每1 ps取1幀,模擬時長200 ps共取得200幀。其中部分反應次數統計如表4所示。蒽二聚物分子數量隨模擬時間的變化曲線如圖10所示。
由表4可知,反應次數最多的是2個蒽二聚物分子縮合成·C56H36(D1)。但·C56H36屬于熱穩定性較差的自由基,很快發生逆反應(D2),因而對后續反應影響很小。對后續反應影響較大的主要是分子間氫轉移反應(D3)。氫轉移引發生成的自由基·C28H17可能與周圍二聚物分子結合生成·C56H35(D4)。由圖10可觀察到,蒽二聚物分子數量隨著模擬時間的增長而逐漸減少,從100個減少到80個,總共消耗了20個分子,模擬200 ps時轉化率為20%。
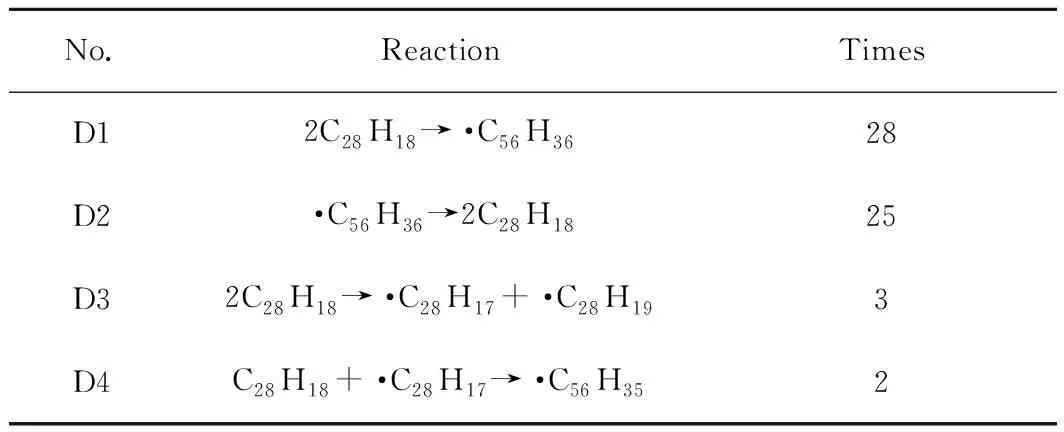
表4 蒽二聚物模型模擬過程中部分反應統計Table 4 Statistics of partial reactions during the simulation
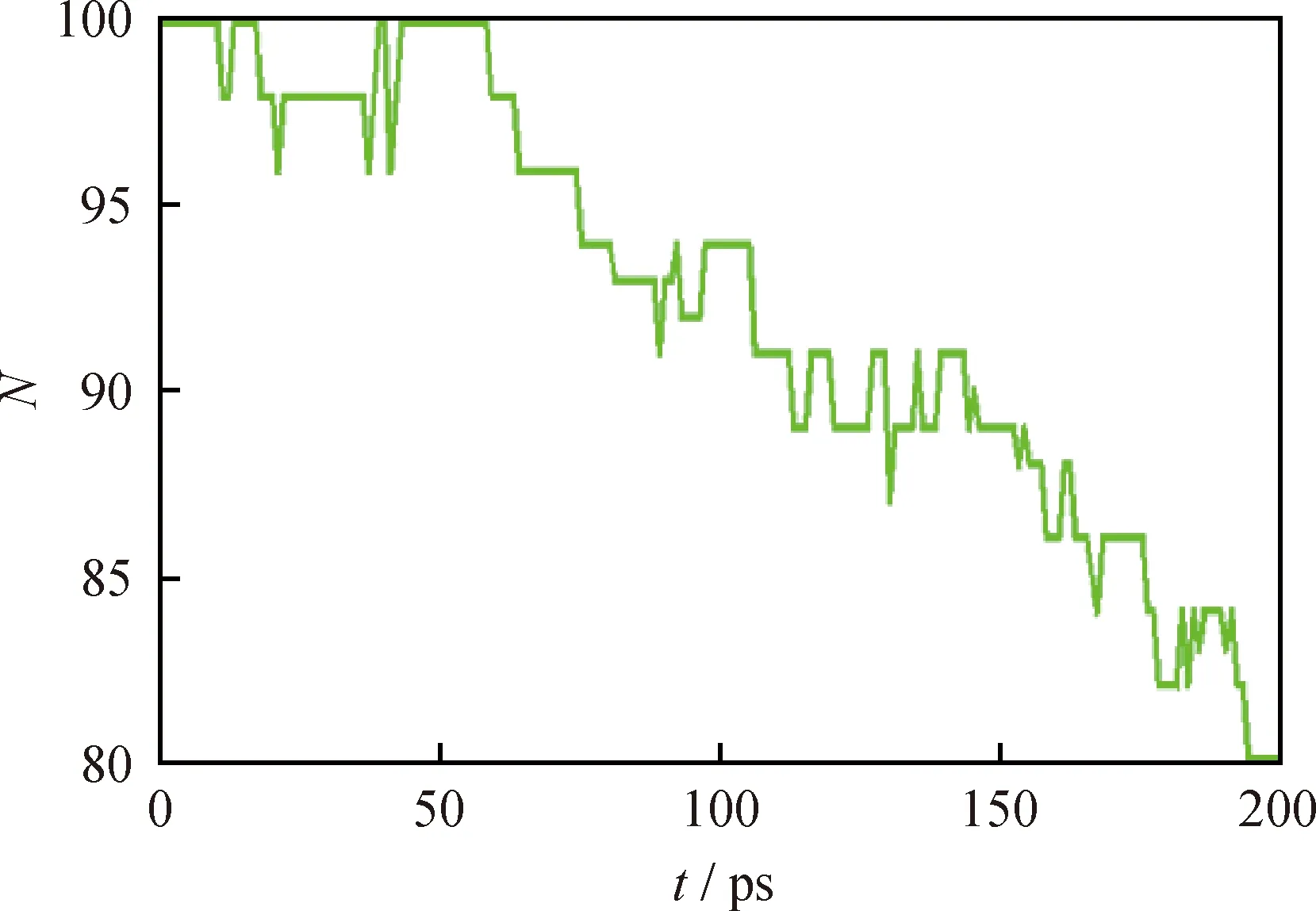
圖10 蒽二聚物分子數量隨模擬時間的變化曲線Fig.10 Molecular number of anthracene dimerversus simulation time
根據混合模型反應分子動力學結果可以觀察到蒽二聚物的反應路線,如圖11所示。由圖11可知,蒽二聚物先通過氫轉移反應引發生成·C28H17自由基。然后·C28H17與周圍的二聚物分子結合生成四聚體自由基。
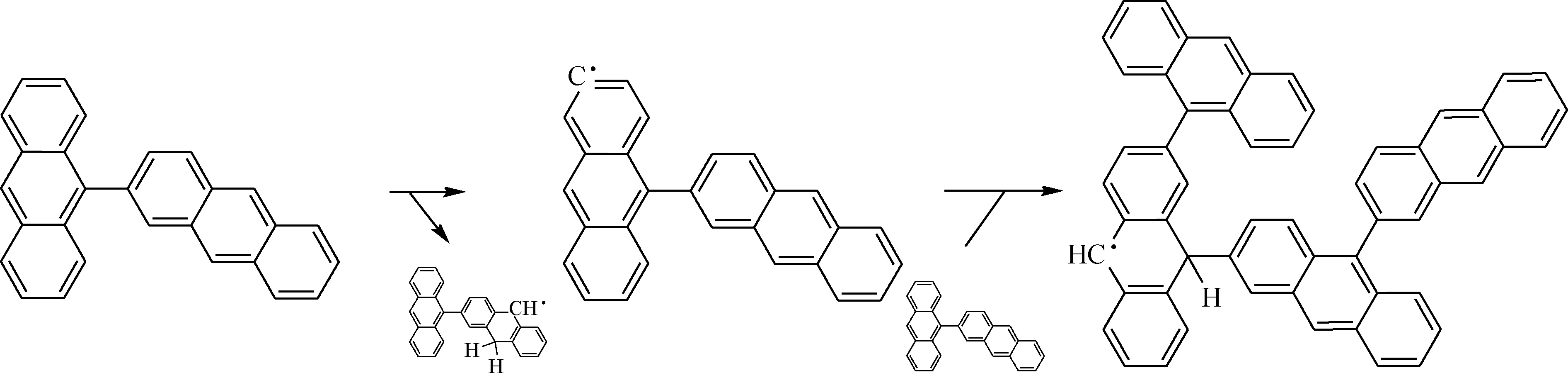
圖11 蒽二聚物模型反應路線Fig.11 Reactive route of dimer model in reactive molecular dynamics simulation
2.4 蒽熱生焦初期的反應路線
根據以上反應分子動力學模擬結果,可以推測蒽在熱生焦初期的反應路線,如圖12所示。由圖12可知:蒽在熱生焦初期,首先通過氫轉移反應引發生成自由基·C14H9;自由基·C14H9與周圍的蒽分子結合生成自由基·C28H19;·C28H19被周圍的蒽分子奪去1個氫原子生成較為穩定的二聚物C28H18;二聚物C28H18通過氫轉移反應生成二聚體自由基·C28H17;·C28H17與周圍的二聚物分子反應,可能生成三聚物C42H26,也可能生成四聚體自由基·C56H35。
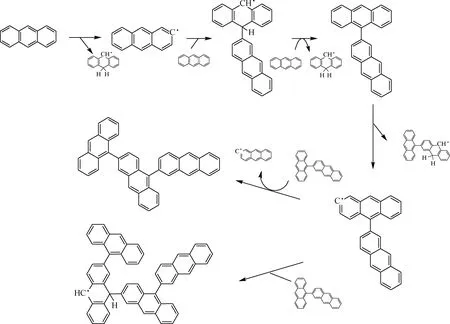
圖12 蒽熱解生焦初期的反應路線Fig.12 Reactive route of anthracene at the initial stage of coking
3 結 論
通過量子化學計算蒽分子結構性質發現,9號位碳的Mülliken凈電荷相比1號位與2號位較多,活性最強,易發生脫氫反應或者氫轉移反應生成自由基。
采用反應分子動力學模擬方法模擬蒽熱生焦初期的反應過程,推斷出蒽熱生焦初期的主要反應歷程:蒽在熱生焦初期,首先通過氫轉移反應引發生成自由基·C14H9;·C14H9與周圍的蒽分子結合生成自由基·C28H19;·C28H19被周圍的蒽分子奪去1個氫原子生成二聚物C28H18;二聚物C28H18通過氫轉移反應生成二聚體自由基·C28H17;·C28H17與周圍的蒽二聚物分子反應,可能生成三聚物 C42H26,也可能生成四聚體自由基·C56H35。重復進行脫氫縮合反應,稠環芳香核隨之增大,最終生成大平面稠環分子。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
網絡安全與數據管理(2022年1期)2022-08-29 03:15:20
導航定位學報(2022年4期)2022-08-15 08:27:00
中學生數理化·中考版(2022年8期)2022-06-14 06:55:24
新世紀智能(數學備考)(2021年9期)2021-11-24 01:14:36
成都醫學院學報(2021年2期)2021-07-19 08:35:14
新世紀智能(數學備考)(2020年9期)2021-01-04 00:25:14
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
光學精密工程(2016年6期)2016-11-07 09:07:19
