改進的動態PPI網絡構建與蛋白質功能預測算法
2020-12-16 02:41:08羅愛靜瞿昊宇許家祺
計算機工程 2020年12期
李 鵬,閔 慧,羅愛靜,瞿昊宇,伊 娜,許家祺
(1.中南大學湘雅三醫院,長沙 410006; 2.湖南中醫藥大學 信息科學與工程學院,長沙 410208; 3.醫學信息研究湖南省普通高等學校重點實驗室(中南大學),長沙 410006; 4.湖南信息職業技術學院 軟件學院,長沙 410200)
0 概述
隨著人類基因組計劃以及多個物種全基因組測序工作的完成,目前生命科學研究的重點已經轉變為蛋白組學[1]。蛋白質是指由多種氨基酸按照某一規律采用多肽鍵所構成的一種多分子化合物,其是生物體中細胞的重要成分,也是生物體完成生命活動最重要的物質基礎[2]。一個生物體內所有蛋白質的相互作用構成了蛋白質相互作用網絡(Protein-Protein Interaction Network,PPIN),簡稱蛋白質網絡[3]。值得注意的是,蛋白質之間的相互作用是動態的,它會隨著時間環境、蛋白質的存在和降解、細胞的不同生理狀態等因素的變化而變化。但由于PPIN本身的復雜性、可利用蛋白質相互作用數據的不完全性和噪聲等諸多因素,準確且高效地衡量蛋白質相互作用的動態性還存在很多挑戰[4],這也直接限制了PPIN領域內其他問題(如復合物挖掘[5]、關鍵蛋白識別[6]、網絡比對[7]等)的研究進展。
文獻[8]從表達動態性、多狀態下表達及相關性變化和時空動態變化3個角度討論了動態蛋白質網絡的構建問題,在此基礎上介紹動態蛋白質網絡在復合物識別、疾病基因檢測等方面的應用,并指出未來動態蛋白質網絡所面臨的挑戰。文獻[9]考慮到酵母物種中蛋白質的基因表達具有時間周期性這一特性,將PPI網絡數據和時間序列基因表達數據相結合構建動態蛋白質交互網絡(Dynamic Protein Interaction Network,D-PIN),并提出一種蛋白質功能預測方法。該文主要通過基于時間的采樣來構建D-PIN,但對不同物種而言,如何合理地選擇一個合適的時機進行采樣仍缺乏理論指導。文獻[10]針對蛋白質功能標簽數量龐大且標簽關聯性較高的特點,提出一種基于布爾矩陣分解的蛋白質功能預測框架PFP-BMD,然而該框架在降低數據噪聲影響方面的效果欠佳。文獻[11]提出一種基于多關系網絡中關鍵功能模塊挖掘的蛋白質功能預測算法PEFM。該算法以高內聚低耦合的原則尋找關鍵功能模塊,并利用這些功能模塊中的鄰居蛋白質信息來注釋未知蛋白質的功能。然而由于需要在多個關系網絡中進行查找,一旦蛋白質之間的相互作用發生改變(如蛋白質降解),則預測效果直線下降,不適用于動態蛋白質網絡。文獻[12]針對現有蛋白質功能預測方法預測精度不高、易受數據噪聲影響等問題,提出一種基于機器學習的蛋白質功能預測方法HPMM,主要采用層次聚類、主成分分析和多層感知器等技術來實現功能預測。然而該方法在訓練多層感知器過程中需要估計的參數較多,時間復雜度較高,且僅適用于靜態蛋白質網絡。
針對以上方法的不足,本文對動態蛋白質網絡的構建問題進行研究,基于進化圖提出一種改進的動態蛋白質網絡構建算法,在此基礎上設計蛋白質功能預測算法IPA-PF,并通過仿真實驗驗證算法的有效性。
1 動態蛋白質網絡構建算法
由于蛋白質之間的相互作用并不是一成不變的,因此本文采用進化圖[13]對動態蛋白質網絡進行建模。為便于描述,給出建模過程中用到的定義:
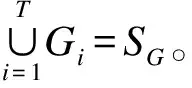
定義2(蛋白質的活性周期) 對于任意給定的一個蛋白質P,如果在一個給定的時間周期T內P的基因表達平均值u(P)都不低于閾值ε,則稱T(P)為P的活性周期。
1.1 動態蛋白質網絡的構建
根據上述定義,動態蛋白質網絡的構建主要包含以下3個步驟:
步驟1根據蛋白質基因表達數據的平均值計算所有蛋白質的活性周期。
步驟2根據所有蛋白質的不同活性周期劃分出多個時間片,擁有相同活性周期的蛋白質屬于同一個時間片。對于處于同一時間片的所有蛋白質,根據它們之間的連接強度構成一個蛋白質子網。
步驟3對步驟2得到的各個時間片的子網,采用進化圖進行建模,最終得到一個全局的動態蛋白質網絡。
1.1.1 活性周期計算
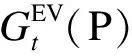
(1)
(2)
進一步地,本文采用F(P)反映蛋白質P基因表達曲線的波動性:
(3)
可以看出,標準差越大,F越小,F的范圍為[0,1]。活性閾值ε的選取參考文獻[14]中提出的3-sigma準則,如下所示:
ε=u(P)×F(P)+(u(P)+3σ(P))×(1-F(P))
(4)
如果在某一時間片Tx內有u(Pi)≥ε,i=1,2,…,k,則認為這k個蛋白質具有相同的活性周期,可用于構建同一個蛋白質子網。通過活性周期的計算可以得到一個關于所有蛋白質活性周期的集合S_T={T1,T2,…,Tk}。本文根據S_T中元素的個數決定劃分出時間片的個數,即構建子網的個數。
1.1.2 蛋白質子網構建
以某一個子網為例來闡述其構建過程,其余子網的構建與此類似。設P_S={P1,P2,…,Pn}表示具有相同活性周期(同一時間片)的所有蛋白質集合,要在這n個蛋白質之間構造一個子網,即要找到n個蛋白質之間的相互作用關系。本文通過考查這些蛋白質之間的連接強度來判斷它們之間是否具有相互作用,如果認為它們之間有相互作用,則在這兩個蛋白質之間添加一條邊。
連接強度主要從兩方面衡量,即直接連接數和間接連接數。直接連接數主要是指兩個蛋白質之間擁有的共同鄰居節點數,如果兩個蛋白質有更多共同鄰居,則表明這兩個節點之間的關系更為緊密,更有可能發生相互作用;間接連接數指兩個蛋白質之間直接相連的邊數和節點的度最小值的比值,它也可以用來衡量蛋白質之間相互作用的強弱。因此,連接強度的定義如下所示:
定義3(連接強度) 蛋白質Pi和蛋白質Pj之間的連接強度JS(Pi,Pj)計算公式如下:
(5)
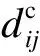
1.2 動態蛋白質網絡模型
動態蛋白質網絡與靜態蛋白質網絡的本質區別在于網絡拓撲因時間、外界環境等因素的動態變化而導致連通性動態變化。如何利用合適的模型來刻畫這種動態性是對蛋白質網絡準確建模的關鍵。考慮到蛋白質的基因表達值具有時間周期性,本文首先將整個蛋白質網絡的運行時間劃分為多個時間片,刻畫出每個時間片內的連通情況,然后利用進化圖的時間演化特性將連續時間片內的多個子圖構建為運行時間內的進化圖模型。
圖1給出了蛋白質網絡工作過程中不同時刻節點相互作用的動態變化情況。其中,頂點是蛋白質,邊表示蛋白質之間的相互作用。假設T1~T4為整個網絡生命周期內任意4個連續的時間片,分別可以構建得到這4個連續時間片內的網絡快照。

圖1 動態蛋白質網絡連續時間片快照Fig.1 Snapshots of continuous time slices ofdynamic protein network
根據定義1,將圖1所示的連續時間片快照建模為進化圖模型。圖1所示時間片快照中的蛋白質(A,B,C,D,E,F,G,H,I,J,K,L)對應于定義1中的頂點集合V,邊集合對應于定義1中的邊集合E,時間序列集合(T1,T2,T3,T4)對應于定義1中的有序時間序列TS。建模過程如下:
1)構造T1時間片內蛋白質網絡連通情況所對應的進化圖子圖G1,并在新出現的每條邊上增加時間序列元素T1。
2)在G1的基礎上累加構造T2時間片內蛋白質網絡連通情況所對應的進化圖子圖G2,并在T2時間片內出現的邊上增加時間序列元素T2。
3)以此類推,直到全部的時間片所對應的進化圖子圖構造完成,得到的進化圖模型如圖2所示。其中,每條邊上的數字序列代表該相互作用存在對應的時間序列,標識該相互作用在第幾個時間片中出現,例如蛋白質A和蛋白質D只在第1個、第2個和第4個時間片內存在相互作用。
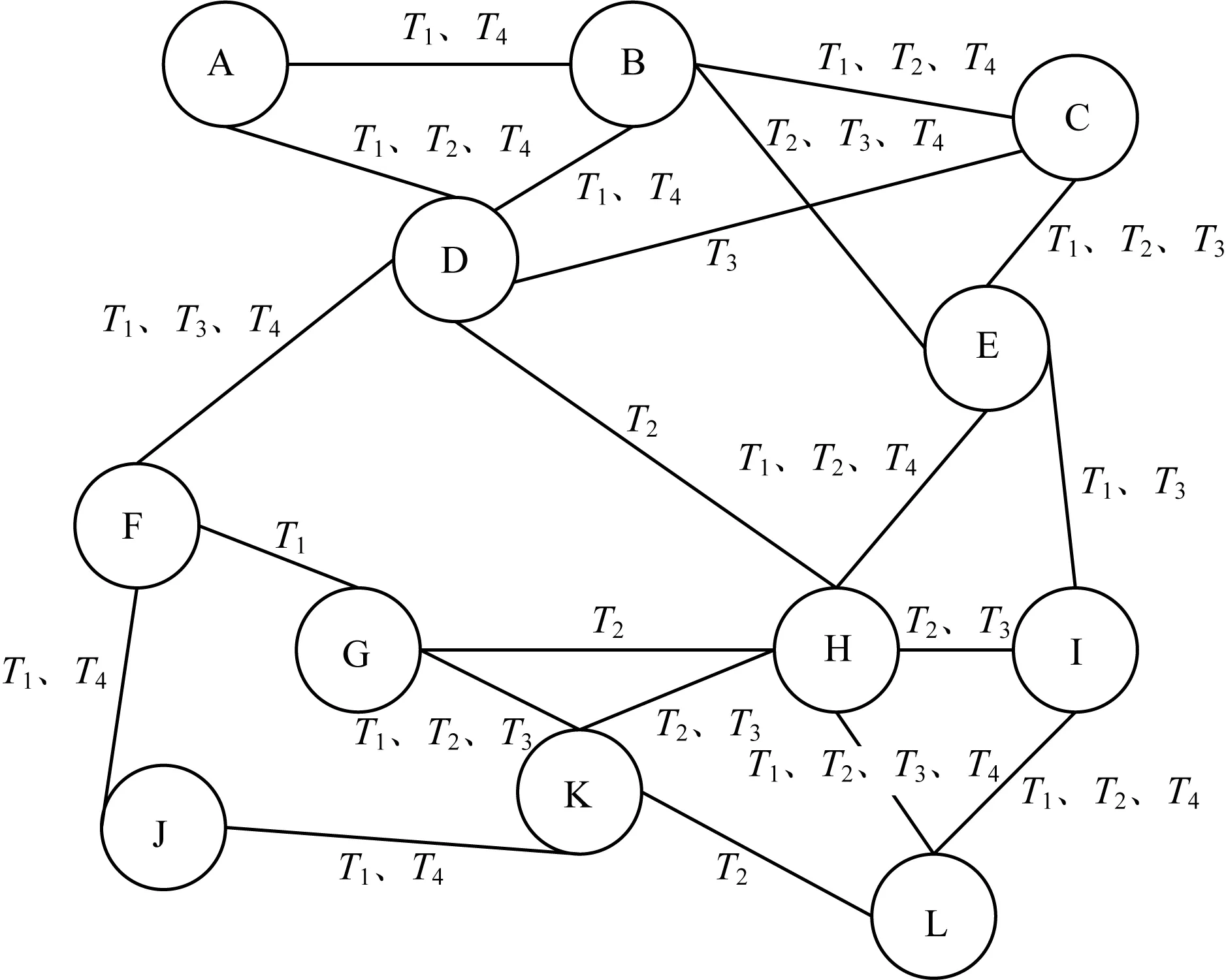
圖2 基于進化圖的動態蛋白質網絡模型Fig.2 Dynamic protein network model based onevolutionary graph
本文提出的動態蛋白質網絡構建算法描述如下:
算法1動態蛋白質網絡構建算法
輸入蛋白質相互作用數據,閾值th,基因表達數據

步驟1根據所有蛋白質的基因表達數據,結合式(1)~式(3)計算所有蛋白質的活性周期T(P),然后對計算結果進行降序排列并采用列表存儲,記為:T(P)=[T1(P),T2(P),…,Tk(P)]。
步驟2根據蛋白質的活性周期構造子網:
ForTi(P),i=1,2,…,kinT(P):
在Ti(P)中計算JS(Pi,Pj);

步驟3重復執行步驟2,直到列表T(P)為空,算法結束。
2 蛋白質功能預測算法
在上文構建得到的動態蛋白質網絡基礎上,提出一種改進的蛋白質未知功能預測算法IPA-PF。首先對待預測功能的蛋白質在T個蛋白質子網中出現的鄰居節點進行統計,然后根據其鄰居蛋白質的功能已知與否,分情況進行處理。
1)如果待預測功能的蛋白質其所有鄰居節點的全部功能或部分功能已知,則根據待預測功能的蛋白質與鄰居蛋白質之間的連接強度來篩選參與功能預測的鄰居蛋白質數目,然后通過計算候選功能得分和排序等操作實現蛋白質的未知功能預測。相關定義及具體過程如下:
定義4(功能關聯得分) 設SG={G1,G2,…,GT}是基于進化圖構建得到的T個蛋白質子網,Gi=(Vi,Ei,ti)。α是一個待預測的功能未知的蛋白質,β是一個功能已知的蛋白質,則β在預測α功能時的功能關聯得分為:
(6)
設NS={P1,P2,…,Pn}是根據式(6)預測α的功能時形成的鄰居蛋白質集合,F={f1,f2,…,fm}是NS集合中所有蛋白質的已知功能集合。設fi是F中某一蛋白質的候選功能,fi的得分為:
(7)
其中,j=1,2,…,m。對NS中所有蛋白質的候選功能根據式(7)的得分進行降序排列,并從中選取前R項功能作為蛋白質α的未知功能列表。本文算法統計NS中每一個蛋白質擁有的功能注釋數量,取其中所有蛋白質的功能注釋數量的最小值作為R的取值。最后,將各個鄰居蛋白質的已知功能注釋的交集作為待預測蛋白質α的功能。例如,對于α的鄰居蛋白質{P1,P2,P3,P4}而言,蛋白質P1擁有功能{f2,f3,f7,f8},蛋白質P2擁有功能{f1,f2,f3,f6},蛋白質P3擁有功能{f2,f3,f5,f9},蛋白質P4擁有功能{f2,f3,f11,f13},因此,可以預測α擁有的功能為{f2,f3}。
2)如果待預測功能的蛋白質其所有鄰居蛋白質節點的全部功能未知,則通過構建一個三層神經網絡[16](包含輸入層、隱藏層和輸出層)模型來進行功能預測,如圖3所示。

圖3 基于三層神經網絡的蛋白質功能預測過程Fig.3 Process of protein function prediction based onthree-layer neural network

本文提出的動態蛋白質網絡蛋白質未知功能預測算法描述如下:
算法2蛋白質未知功能預測算法IPA-PF

輸出未知蛋白的功能注釋
步驟1對于每一個待預測功能的蛋白質α,統計其在SG中出現的鄰居蛋白質節點,記為集合NS={P1,P2,…,Pk}。
步驟2如果NS中蛋白質的全部功能或部分功能已知,則:
1)根據式(6)和式(7)計算NS中所有蛋白質的候選功能得分,并對得分進行降序排列,取前R項。
2)計算NS中所有蛋白質前R項功能的交集,然后轉步驟4。
步驟3如果NS中蛋白質的全部功能未知,則訓練一個神經網絡進行蛋白質功能預測:
1)數據預處理:采用丟棄、填充、替換或去重等操作對蛋白質的特征做歸一化處理。
2)在(0,1)區間內隨機初始化網絡中的所有連接權值和閾值。
3)根據蛋白質的特征,采用累積誤差逆傳播算法[18]進行訓練,得到一個連接權值與閾值確定的三層前饋神經網絡(3-FNN)。
4)采用3-FNN進行蛋白質功能預測。
步驟4輸出未知蛋白質的功能注釋。
3 實驗
實驗利用Python語言實現本文提出的動態蛋白質網絡構建算法和蛋白質未知功能預測算法IPA-PF。為驗證動態蛋白質網絡構建算法的合理性和IPA-PF的有效性,在多個數據集上將IPA-PF算法與目前較為典型的蛋白質功能預測算法D-PIN[9]、PFP-BMD[10]、PEFM[11]和HPMM[12]進行性能比較。在一臺8核16線程的計算機上進行實驗。其中,CPU型號為Intel Core i9-9960X@3.10 GHz,內存為16 GB,操作系統為Ubuntu 16.04 LTS 64位系統,采用GPU加速技術和TensorFlow框架來訓練文中用到的神經網絡,GPU型號為GeForce RTX 2070。
3.1 實驗數據集
本文采用DIP數據集、MIPS數據集、GO數據庫[19]和CYC數據集[20]作為測試數據集。其中,DIP數據集記錄了通過生物實驗測定的蛋白質之間的相互作用,它將來自各種來源的信息相互結合,形成一組單一、一致的蛋白質-蛋白質相互作用。本文使用的DIP數據是DIP20170205版本,選取其中的酵母蛋白質網絡來進行實驗。用UniProtKB/Swiss-Prot[21]對PPI網絡中的蛋白質進行ID轉換,然后去除網絡中自相互作用、重復相互作用及無法轉換的蛋白質后,該網絡中還有4 995個蛋白質和21 554條邊。MIPS數據集源自慕尼黑蛋白質序列信息中心,本文采用和上述相同的方法進行數據預處理,最終得到的相互作用網絡包括4 546個酵母蛋白質和12 319對可靠的相互作用。下載基因本體(Gene Ontology,GO)數據庫的最新版本來測試不同算法在蛋白質功能預測方面的性能。其中包含細胞組件、分子功能和生物過程3個獨立的子本體。為保證功能預測的全面性和高效性,本文保留未被GO術語注釋的蛋白質,并且保留功能注釋數目不超過200個蛋白質的GO Term來進行算法驗證。此外,將CYC2008作為基準數據集來評估蛋白質復合物的識別結果。該數據集中包含408個通過生物方法預測到的蛋白質復合物,每個復合物包含兩個或兩個以上蛋白質。
3.2 評價指標
本文采用以下指標來評價不同算法的性能:
1)查全率、查準率和F-measure值。查全率(Recall)為預測的蛋白質功能與實驗數據集中真實存在的蛋白質功能注釋的最大匹配數目與實驗數據集中真實存在的蛋白質功能注釋總數的比值,查準率(Precision)為預測的蛋白質功能與實驗數據集中真實存在的蛋白質功能注釋的最大匹配數目與實驗測得的蛋白質功能注釋總數的比值,這兩個指標的計算公式如下:
(8)
(9)
其中:ER表示本文算法預測的蛋白質功能;RR表示實驗數據集中真實存在的蛋白質功能注釋;MNM(ER,RR)表示ER和RR之間的最大匹配數目。綜合考慮查全率和查準率兩方面,可得F-measure的計算公式為:
(10)
2)魯棒性。目前能夠獲得的蛋白質相互作用數據都在一定程度上存在假陽性和假陰性的問題。因此,一個優秀的蛋白質構建算法和功能預測算法應對數據中存在的假陽性和假陰性具有很好的魯棒性。
3)時間開銷。在多個數據集上衡量動態蛋白質網絡構建算法和蛋白質功能預測算法運行所耗費的時間,比較不同算法的運行效率。
3.3 實驗結果與分析
3.3.1 IPA-PF算法與其他算法的比較
為全面分析本文提出的動態蛋白質網絡構建算法和IPA-PF算法的性能,將IPA-PF算法與D-PIN[9]、PFP-BMD[10]、PEFM[11]和HPMM[12]在DIP數據集和MIPS數據集上進行比較。采用十折交叉驗證法進行實驗評估,即將DIP數據集和MIPS數據集分別分成10份,輪流將其中9份作為訓練數據,將1份作為測試數據。為進一步降低實驗誤差,重復進行100次實驗,取其平均值作為最終的結果。表1和表2分別列出了不同算法在DIP數據集和MIPS數據集上的性能比較。

表1 不同算法在DIP數據集上的性能比較Table 1 Performance comparison of different algorithmson DIP dataset

表2 不同算法在MIPS數據集上的性能比較Table 2 Performance comparison of different algorithmson MIPS dataset
從表1和表2的結果可以看出,本文算法在兩種數據集上的查全率和查準率都要優于其他4種算法,并且在DIP數據集上,本文算法的F-measure值較HPMM、D-PIN、PEFM和PFP-BMD分別提高約40%、30%、26%和16%,在MIPS數據集上,本文算法的F-measure值較HPMM、D-PIN、PEFM和PFP-BMD分別提高約39%、26%、25%和11%,主要原因如下:
1)本文算法在構建動態蛋白質網絡的過程中考慮了蛋白質基因表達的活性周期,能夠更好地模擬蛋白質“合成-降解-凋亡”這一個生物過程,避免了網絡構建的片面性。
2)通過引入連接強度這一概念,從物理位置上對蛋白質節點之間的相互作用進行評價,從而有效過濾了蛋白質相互作用數據中所隱含的假陽性和假陰性。
3)在未知蛋白的功能預測方面,本文對D-PIN算法的不足之處進行了改進,對待預測蛋白質節點的鄰居蛋白質節點分情況(有功能注釋/無功能注釋)進行處理,并考慮蛋白質的多種特征來訓練神經網絡進行功能預測,解決了當鄰居蛋白質節點的功能集合全部未知時無法進行預測這一難題,因此,本文算法能夠更全面地預測蛋白質的未知功能。
3.3.2 參數th對蛋白質復合物識別性能的影響分析
在動態蛋白質網絡構建過程中,參數th對于衡量兩個蛋白質之間是否具有相互作用起到關鍵作用,下面以CYC2008數據集為實驗對象,測試th取不同數值時構建出的網絡在蛋白質復合物上的識別性能,選取兩種典型的蛋白質復合物識別算法(MPC-TPW[22]和DPC-NADPIN[23])來分析本文構建網絡算法的可靠性,實驗結果如圖4所示。可以看出:隨著th取值增大,MPC-TP算法和DPC-NADPIN算法的F-measure值呈現不斷增加的趨勢,這表明兩種算法能夠準確識別的蛋白質復合物數量越來越多;但在th取值達到0.7之后,MPC-TP算法和DPC-NADPIN算法的性能趨于穩定,這表明本文提出的動態蛋白質網絡構建算法對于輸入參數不敏感,能夠應用到不同的蛋白質復合物識別算法中。

圖4 不同蛋白質復合物識別算法的參數敏感性比較Fig.4 Parameter sensitivity comparison of differentprotein complex recognition algorithms
3.3.3 魯棒性分析
測試IPA-PF算法對于包含假陰性和假陽性的蛋白質相互作用數據的魯棒性。以DIP數據集為測試用例,在實驗中通過隨機增加和刪除一定比例的邊來模擬蛋白質網絡的假陽性和假陰性。其中:假陽性是指能夠被實驗技術檢測到但在細胞中并不存在的蛋白質相互作用;假陰性是指不能被實驗技術檢測到但在細胞中確實存在的蛋白質相互作用。以每20個百分點為一個間隔,隨機地增加邊的比例從20%到100%,共得到5組數據,從這些具有較高假陽性的數據中識別蛋白質復合物,得到IPA-PF算法的查全率和查準率,如圖5所示。可以看出,隨著假陽性的增強,IPA-PF算法預測蛋白質功能的查全率基本保持不變,而查準率有輕微下降,這表明IPA-PF算法具有較強的抗噪能力,能夠應對那些被算法檢測得到但在數據集中并不存在的蛋白質相互作用。

圖5 數據包含假陽性時IPA-PF算法的性能指標Fig.5 Performance indexes of IPA-PF algorithmwith false positive data
以每20個百分點為一個間隔,隨機地刪除邊的比例從15%到90%,共得到6組數據,重復上述工作,得到IPA-PF算法的查全率和查準率,如圖6所示。可以看出:當刪除邊的比例小于45%時,IPA-PF算法預測蛋白質功能的查全率和查準率基本保持不變;在刪除邊的比例超過40%后,IPA-PF算法的性能開始呈現直線下降趨勢,這是因為隨著假陰性的增強,數據集中那些未被IPA-PF算法檢測到但又真實存在的相互作用會被大量刪除,理論上會使算法能夠預測的蛋白質功能數量急劇減少,而IPA-PF算法反映在查全率和查準率上的變化就是這兩種指標直接降低,這也恰好驗證了IPA-PF算法對于假陰性具有較好的魯棒性。

圖6 數據包含假陰性時IPA-PF算法的性能指標Fig.6 Performance indexes of IPA-PF algorithm withfalse negative data
3.3.4 不同算法的效率分析
為進一步衡量本文算法的優越性,在上述實驗環境下對不同蛋白質功能預測算法的時間開銷進行測試。以DIP數據集和MIPS數據集作為測試用例,表3給出了不同算法在進行蛋白質未知功能預測時的運行時間。可以看出,IPA-PF算法在兩種數據集上的運行時間均不超過11 s,低于D-PIN、PEFM和HPMM算法,略高于PFP-BMD算法。但通過上文的實驗分析結果可知,IPA-PF算法的預測質量遠超其他預測算法。從性能折中的角度來看,以目前計算機的算力而言,在保證蛋白質功能預測準確性的前提下,犧牲算法的部分效率完全是可以接受的。總體而言,本文提出的IPA-PF算法具有較高的運行效率,可適用于大規模的蛋白質網絡。

表3 不同算法的運行時間比較Table 3 Running time comparison ofdifferent algorithms s
4 結束語
蛋白質相互作用網絡是目前蛋白組學的研究熱點。針對現有蛋白質網絡構建和功能預測方法存在的不足,本文提出一種基于進化圖的動態蛋白質網絡構建算法,在此基礎上設計一種新的蛋白質功能預測算法,并在多個公開的生物數據庫上驗證算法的有效性。本文研究有利于從微觀層面解釋細胞內蛋白質之間的復雜關系,為生物學和醫學領域研究者理解生命復雜網絡的內在組織和生物過程提供了新的途徑,并可用于藥物標靶設計、疾病診治和預測等多個方面。下一步將分析影響動態蛋白質網絡構建的諸多因素,并采用深度學習技術對關鍵蛋白質的識別進行建模,設計基于圖卷積神經網絡的關鍵蛋白質識別算法。
猜你喜歡
中華詩詞(2022年6期)2022-12-31 06:41:24
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
中國科技論壇(2017年7期)2017-07-25 08:49:53
媽媽寶寶(2017年2期)2017-02-21 01:21:24
國際漢語學報(2016年1期)2017-01-20 08:21:20
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
中國中醫藥現代遠程教育(2014年22期)2014-03-01 04:32:55
