UPLC-MS/MS法測定金沙江水域魚體中卡巴氧及喹乙醇代謝物
2020-12-14 11:25:34孫卓然田金鳳尚遠宏
中國釀造 2020年11期
關鍵詞:方法
孫卓然,田金鳳,尚遠宏
(1.呼和浩特市疾病預防控制中心,內蒙古 呼和浩特 010070;2.攀枝花學院 醫學院,四川 攀枝花 617000)
水產品是人類食用蛋白質的重要來源之一,而水產養殖中使用的藥物殘留將會對行業發展和人體健康產生不利的作用。卡巴氧(carbadox)和喹乙醇(olaquindox)屬于喹啉類藥物,由于其具有明顯的抗菌和促生長作用,曾在水產養殖業中應用,但后期證明有較強的毒副作用,已被不同程度地限制或禁止使用[1-2]。3-甲基-喹惡啉-2-羧酸(3-methylquinoxaline-2-carboxylic acid,MQCA)和喹惡啉-2-羧酸(quinoxaline-carboxylic acid,QCA)分別是喹乙醇和卡巴氧在靶動物體內的代謝物,被定為殘留檢測的標示物[3]。卡巴氧和喹乙醇本身具有潛在的致癌、致畸作用,二者的代謝物也可能帶來健康風險,因此多個國家對二者及其代謝產物QCA和MQCA制定了殘留監控的限量標準[4-6]。同時,我國現執行的水產行業標準SC/T 3019—2004《水產品中喹乙醇殘留量的測定液相色譜法》和農業部1077號公告-5-2008有水產品中喹乙醇殘留量分析的高效液相色譜方法,但處理方法的靈敏度低,殘留分析的可靠性低;且尚未見直接控制水產品自身組織中卡巴氧的檢測標準,而只是有對其飼料中的卡巴氧殘留檢測的標準。因此,探討并建立檢測水產品中喹乙醇和卡巴氧殘留方法,對于完善和改進水產品中2種喹啉類藥物殘留標準和方法具有實際意義。
目前,檢測在動物源食品中QCA和MQCA的方法文獻報道較多,最常用的有液相色譜法[7-8]、免疫法[9-10]和液相色譜-質譜聯用法[11-14],但對魚類檢測的應用較少。本研究在前人研究工作的基礎上,明確了區域定位和增加且細化了檢測品種,極大提高了方法的靈敏度。實驗以金沙江攀枝花江段6種常見商品魚為研究為對象,適當改進了樣品前處理方法,建立了魚組織中QCA和MQCA殘留的超高效液相色譜串聯三重四極桿質譜(ultra-performance liquid chromategr-aphy-triple quadrupole mass spectrometry,UPLC-MS/MS)檢測方法,為魚組織中QCA和MQCA殘留的評估檢測提供參考。
1 材料與方法
1.1 材料與試劑
1.1.1 材料
樣品來源于金沙江攀枝花江段6種常見魚(羅非魚、草魚、鯉魚、鰱魚、鯽魚、鯰魚),且以羅非魚作為方法驗證樣品,本地市場或養魚場隨機購得,鮮活的羅非魚、鯉魚等水產品去鱗、去皮、沿背脊取肌肉,充分絞碎,均質,-20 ℃以下避光保存。
1.1.2 化學試劑
蛋白酶(5 U/mg)、Tris堿(HOCH2)3CNH2(純度98.5%):美國Sigma公司;喹惡啉-2-羧酸(純度98.9%)、3-甲基喹惡啉-2-羧酸(純度98.1%)、喹惡啉-2-羧酸-d4(純度99.5%,QCA-d4)標準品:德國Dr.Ehrenstorfer GmbH公司;甲酸、甲醇、乙酸乙酯、乙腈(均為色譜純):美國Fisher公司;鹽酸、乙酸、氯化鈣等其他試劑為國產分析純。
1.2 儀器與設備
Agilent 6460A超高效液相色譜-串聯四級桿質譜儀、Bond Elut Plexa PAX 固相萃取柱(200 mg/6 mL,使用前依次用6 mL甲醇、6 mL水活化,保持柱體濕潤):美國Agilent 公司;Vortex 4渦旋混合器:比利時LMS公司;Milli-Q IQ7000超純水器:美國Millipore公司;Nevap-45氮吹儀:美國Organomation公司;FA2014B電子天平:上海越平公司;1-14k離心機:美國Sigma公司。
1.3 方法
1.3.1 標準溶液的配制
分別稱取0.0050 g喹惡啉-2-羧酸、喹惡啉-2-羧酸-d4(同位素內標,QCA-d4)、3-甲基喹惡啉-2-羧酸標準品,分別用甲醇溶解并定容至10 mL,質量濃度均為0.5 mg/mL,-20 ℃避光保存。其他濃度標準溶液用甲醇定量混合或逐級稀釋即得。
1.3.2 樣品前處理
樣品提取:分別稱取6種魚的均質樣品5.0 g于50 mL離心管中,加入100 μL 100 μg/mL QCA-d4溶液、8 mL 0.2 mol/L(pH=9.6)Tris緩沖溶液和300 μL 10 mg/mL蛋白酶水溶液,渦旋混勻,置于空氣浴恒溫搖床中47 ℃酶解18 h,取出樣品放置至室溫,再加1 mL濃鹽酸,振搖混勻,4 ℃、9 000 r/min離心5 min。取上層清液加入6 mL乙酸乙酯,渦旋1 min,4 ℃、9 000 r/min離心5 min,再重復乙酸乙酯步驟1次,合并兩次上清液。上清液于35 ℃下氮吹濃縮至近干,殘渣用10 mL 20%甲醇水溶解,待凈化。
樣品凈化:將樣品提取液轉移至經活化的PAX柱。依次用6 mL水、6 mL 2%氨水、6 mL甲醇和6 mL 0.5%乙酸-甲醇淋洗PAX固相萃取柱。然后將PAX柱抽干,用6 mL 2%甲酸-甲醇溶液洗脫目標化合物,收集洗脫液,35 ℃下氮吹濃縮至干,用1.0 mL 20%甲醇-水溶液溶解殘渣,過0.22 μm濾膜后,備用。
1.3.3 儀器條件
液相色譜條件:流動相:0.1%的甲酸水溶液-乙腈,梯度洗脫;流速:0.25 mL/min;進樣體積:5 μL;色譜柱:ZORBAX SB-C18(2.1 mm×100 mm,1.8 μm);柱溫:40 ℃;洗脫程序:0~6 min,乙腈由5%線性升至25%;6~7 min,25%乙腈;7.0~7.1 min,乙腈由25%線性降至5%。
質譜條件:電子電離(electronic ionization,EI)源;掃描方式為QCA、MQCA、QCA-d4為正離子掃描;多反應監測(multiple-reaction monitoring,MRM);電噴霧電壓:4 000 V;輔助氣(auxiliary gas flow,AUX)流速:11 L/min;輔助氣溫度:350 ℃。
1.3.4 標準曲線的繪制及檢出限的測定
配制好的QCA和MQCA標準儲備液用甲醇稀釋成400 μg/L混合標準溶液,用空白羅非魚基質提取液將混合標準溶液稀釋成2.0 μg/L、4.0 μg/L、10.0 μg/L、20 μg/L、40 μg/L、80 μg/L和100 μg/L系列質量濃度的混合標準溶液分別進行測定。以目標化合物的質量濃度(x)為橫坐標和其峰面積(y)為縱坐標,繪制工作曲線。以3倍信噪比(S/N>3)和10倍信噪比(S/N>10)分別確定檢出限(detection limit,LOD)與定量限(limit of quantitation,LOQ)[15-17]。
2 結果與分析
2.1 樣品提取與凈化
魚肉樣品含有15%~20%的蛋白質和5%左右的脂肪[18],而喹噁啉類藥物在動物源性食品中的殘留具有基質復雜且固體基質內源性干擾較大、含量低(痕量)的特點[19],對藥物分析的干擾很大,因而必須進行樣品前處理。QCA和MQCA與原藥的結構和性質差別較大,為有機強酸,極性較強,在動物體內通過共價鍵與一些氨基酸(或蛋白質)結合,需要利用酶水解為游離態,再進行提取,且減少了雜質干擾。酶解液經鹽酸酸化,乙酸乙酯提取,可有效沉淀蛋白質,消除代謝活性物質及終產物對提取的影響,乙酸乙酯洗脫液的雜質較少,提高了絕對回收率。同時,實驗比較了不同規格(60 mg/3mL、200mg/6mL、500mg/6mL)的PAX小柱,直管型柱中填料量和體積過小(60 mg/3 mL)存在回收率低和柱飽和現象,過大又存在浪費特異性固相萃取材料不經濟現象,最后選擇PAX(200 mg/6 mL)柱回收率較高,可有效除去干擾物質,滿足實驗要求。
2.2 儀器條件的優化
在MQCA和QCA 殘留分析中使用的流動相為0.1%甲酸-乙腈體系,因甲酸可以為化合物提供必需的質子來源,提高其離子化效率。在ESI+模式下,優化碰撞能量,MQCA和QCA經過兩級碰撞掃描后,產生不同強度的離子碎片,以豐度最大的m/z 145、m/z133和m/z 129作為3種化合物的定量離子,MQCA 和QCA的MRM色譜圖見圖1,定量離子色譜圖見圖2,3種化合物的質譜分析參數見表1。
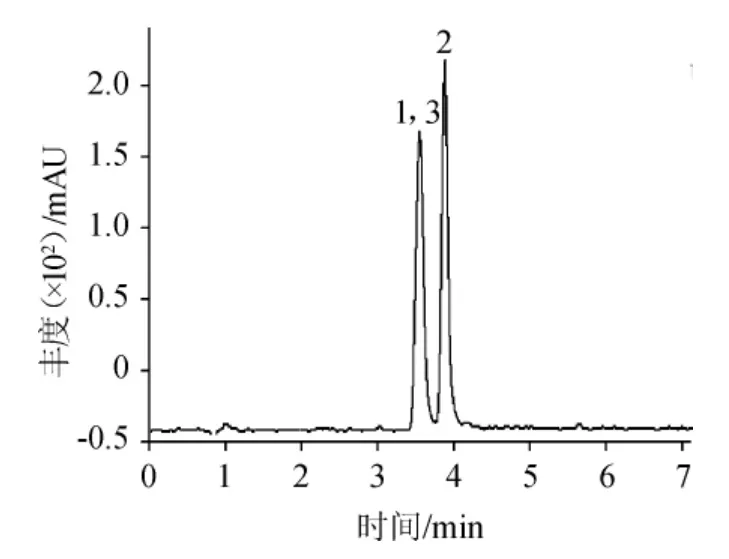
圖1 混合標準溶液的UPLC-MS/MS分析總離子流色譜圖Fig.1 Total ion chromatogram of mixed standard solution analysis by UPLC-MS/MS
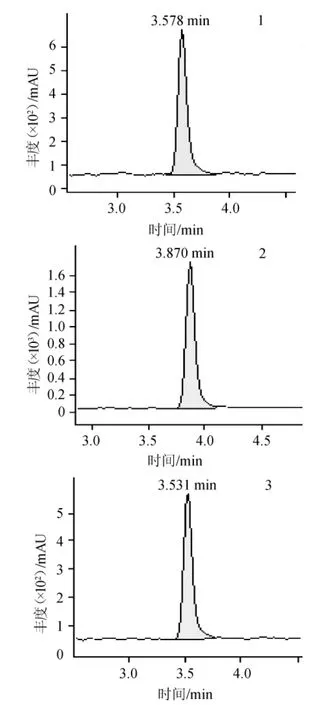
圖2 3種化合物的定量離子色譜圖Fig.2 Quantitative ion chromatogram of 3 compounds
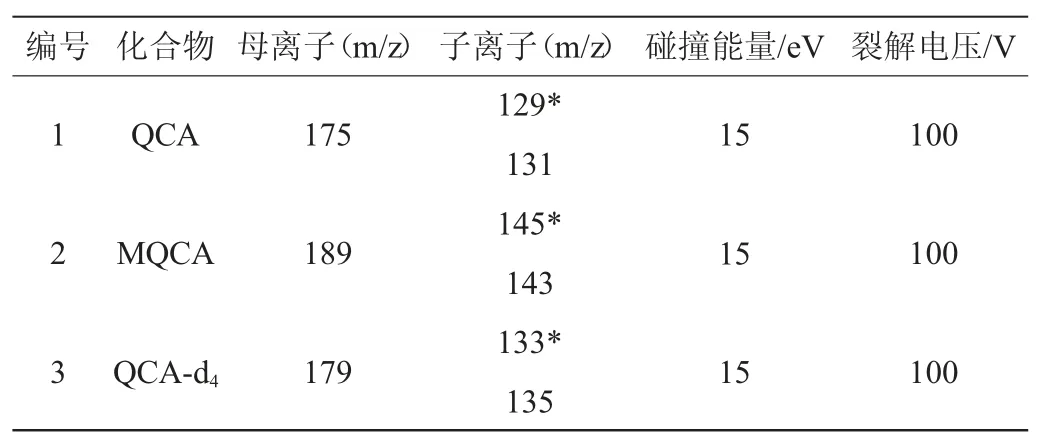
表1 多反應監測模式下3種獸藥的質譜參數Table 1 MS parameters of 3 veterinary drugs under MRM mode
由圖1和圖2可知,MQCA和QCA經色譜分離流出的組分不斷進入質譜,質譜連續掃描進行數據采集,每一次掃描得到一張質譜圖,將每一張質譜圖中所有離子強度相加,得到一個混合標準溶液總的離子流強度。同時,定量離子色譜圖反映了色譜峰的定性(目標化合物的特征離子),峰的一致性測定。
2.3 方法的線性關系及檢出限
向空白羅非魚基質溶液中加入系列混合標準溶液和內標溶液,以峰面積對應的濃度進行線性回歸,結果見表2。由表2可知,其線性關系良好,相關系數(R)>0.999。定量限(LOQ)為0.2 μg/kg,檢出限(LOD)為0.05 μg/kg。結果表明,方法的靈敏度能滿足檢測要求,并優于已有標準[6]和文獻[3,14]。
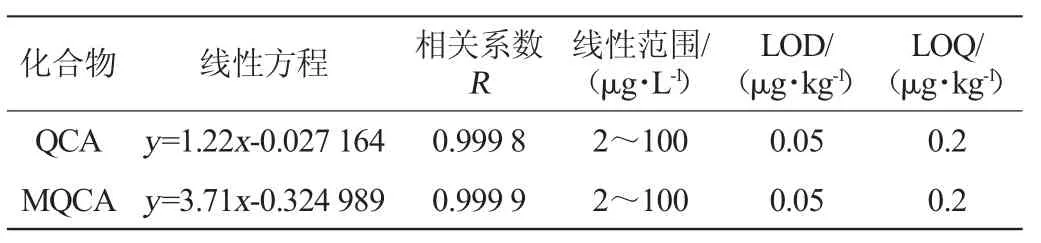
表2 方法的線性回歸方程、相關系數和檢出限Table 2 Linear regression equations,correlation coefficients and detection limits of method
2.4 方法的回收率與精密度
空白的羅非魚樣品分別添加不同濃度的標準液各6份,搖勻、靜置后按1.3.2方法處理并進行加標回收率及精密度試驗,計算平均回收率和相對標準偏差(relative standard deviation,RSD),結果見表3。由表3可知,各添加水平的回收率為91.4%~100.4%,精密度試驗結果RSD為6.3%~11.5%,方法的準確度與精密度均滿足藥物殘留分析的要求[20]。
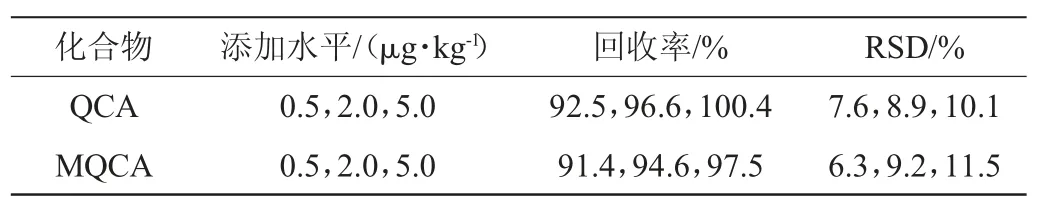
表3 加標回收率和精密度試驗結果Table 3 Results of adding standard recovery rate and precision tests
2.5 實際樣品分析
目前,我國現行標準對水產品中喹乙醇代謝物MQCA的最大殘留限量為4 μg/kg[6],還沒有對卡巴氧代謝物QCA制定最大殘留限量。按試驗方法對金沙江攀枝花江段的30份6種常見商品魚(羅非魚、草魚、鯉魚、鰱魚、鯽魚、鯰魚)進行分析,其QCA和MQCA的殘留量均低于方法的檢出限,均未檢出。
3 結論
羅非魚、草魚、鯉魚、鰱魚、鯽魚、鯰魚作為市場流通的常見水產品,其QCA和MQCA殘留也引起越來越多的關注。雖然我國現行標準僅對水產品中喹乙醇代謝物MQCA殘留量有限定[6],但由于魚體中QCA和MQCA殘留對人類存在健康風險,非常有必要對魚體中的卡巴氧及喹乙醇代謝物殘留進行檢測和監控。本研究采用同位素內標法定量,UPLC-MS/MS法分別測定金沙江攀枝花江段6種常見商品魚中QCA和MQCA的殘留量均未檢出,本方法靈敏度較好,回收率和重現性符合檢測要求,達到了QCA和MQCA殘留分析的要求,為食品安全風險監測魚類等水產品中藥物殘留的檢測方法和限量標準的制定提供了一定的參考依據。
猜你喜歡
中老年保健(2021年9期)2021-08-24 03:52:04
河北畫報(2021年2期)2021-05-25 02:07:46
中學生數理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
兒童繪本(2020年5期)2020-04-07 17:46:30
兒童故事畫報(2019年5期)2019-05-26 14:26:14
Coco薇(2016年2期)2016-03-22 02:42:52
山東青年(2016年1期)2016-02-28 14:25:23
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
小雪花·成長指南(2015年4期)2015-05-19 14:47:56
