雜原子修飾多孔鐵鈷氮碳基氧還原催化劑的電化學性能分析
2020-12-06 10:41:23陳英楠崔勁松劉恩紅柳傲雪韓策楊國程
分析化學 2020年11期
陳英楠 崔勁松 劉恩紅 柳傲雪 韓策 楊國程

摘?要?受限于商用鉑基催化劑穩定性差和生產成本高等缺點,燃料電池以及相關綠色能源的推廣進展緩慢。因此,研發高效、廉價的替代型氧還原電催化劑,是電催化領域的研究熱點。本研究以催化活性較好的鐵鈷氮碳基材料為基礎,通過外加雜原子的方法,制備了一系列雜原子修飾多孔鐵鈷氮碳基氧還原催化劑。通過對所制備材料的組成和形貌進行表征,并結合其在堿性電解質中電催化氧還原的活性、穩定性和抗甲醇能力,探究了雜原子修飾與材料催化性能之間的潛在規律。所制備的FeCo/NFC催化劑,在堿性條件下半波電位為0.84 V,在0.5 V處電子轉移數為3.85,HO2中間產物產率為7.61%,表明此催化劑具有與商業鉑碳相近的催化效果和近四電子的催化選擇性。此外,FeCo/NFC催化劑還具有優良的催化穩定性和抗甲醇能力,作為商業鉑碳的替代品,在燃料電池領域具有良好的應用潛力。
關鍵詞?雜原子修飾; 過渡金屬-氮-碳; 氧還原反應; 電催化劑
1?引 言
為了緩解全球性的氣候變化并解決迫在眉睫的能源枯竭問題,對可持續清潔能源轉換設備的探索勢在必行[1]。近年來,燃料電池由于其高效性和清潔環保的特點,成為一種極具潛力的動力來源。然而,燃料電池陰極緩慢的氧氣還原反應(ORR)是限制其進一步推廣使用的瓶頸[2]。目前,高活性的鉑基材料是最有前途的ORR催化劑,但其高成本、抗甲醇能力差以及易于氧化等缺點,導致使用鉑基催化劑組裝燃料電池的研究和應用的進展緩慢[3]。因此,研發高效、穩定的非貴金屬ORR催化劑成為燃料電池研究領域的當務之急。
對于非鉑ORR催化劑的研究已有大量的文獻報道,其中主要包括過渡金屬基材料和雜原子摻雜碳基材料[4,5]。對于過渡金屬基材料而言,過渡金屬-氮-碳基催化劑是此類材料中最具開發潛質的材料,例如鐵氮碳基和鈷氮碳基材料[6,7]。這主要受益于過渡金屬本身特殊的電子結構和氮摻雜碳對金屬周圍電子排布的誘導作用[8]; 同時,過渡金屬在熱解過程中對碳基質的石墨化作用,可以提升材料的電子傳導能力,并增加催化中心的耐受性,可改善材料的活性和穩定性[9,10]。而對于雜原子摻雜碳材料而言,摻雜的雜原子與碳基質之間的電負性差異會誘發材料電荷重排,從而改善材料與氧氣分子的結合能力,這通常被認為是此類材料ORR活性提升的首要原因[11]。同時,由于不同雜原子之間存在的相互作用,在單摻雜碳基材料中引入其它雜原子被證明也可以顯著提升催化劑的ORR活性[12]。
本研究通過簡單的熱解方式制備了一系列雜原子修飾多孔鐵鈷氮碳基氧還原催化劑。通過分別引入硼、磷和氟,制備的雜原子修飾多孔鐵鈷氮碳基氧還原催化劑(FeCo/BNC、FeCo/NPC和FeCo/NFC)展現出相較于FeCo/NC更優異的催化性能,這不僅是由于引入了雜原子對金屬-氮-碳催化位點的修飾作用,也因材料自身具有較高的比表面積、豐富的孔道結構以及均勻豐富的活性位點。研究結果表明,引入不同的雜原子,可從不同方面改進FeCo/NC的ORR活性:硼的修飾可以有效改善材料的催化選擇性,使FeCo/BNC修飾電極上的ORR過程更趨近四電子催化路徑; 磷的修飾則傾向于提高材料的催化活性,FeCo/NPC修飾電極上的ORR過程,電流密度明顯改善; 而氟的修飾則使FeCo/NFC的ORR催化選擇性和活性均有顯著改善。
2?實驗部分
2.1?儀器與試劑
JEM-2100透射電子顯微鏡(TEM,日本日立公司); ASAP 2020氮氣吸附比表面積分析儀(美國Micromeritics公司); ESCALAB 250 X射線光電子能譜(美國賽默飛公司); CHI 760E電化學工作站(上海辰華公司); Model 636旋轉環盤電極旋轉器(美國普林斯頓應用研究公司)。
單氰胺溶液(50%, m/m)、植酸溶液(50%, m/m)、NH4F、Co(NO3)2·6H2O、FeSO4·7H2O、對羥基苯硼酸(分析純,阿拉丁公司); Pt/C(20%, m/m)、Nafion(5%, m/m)均為分析純(美國Sigma-Aldrich公司); HCl(分析純,北京化工廠)。實驗用水為美國Millipore公司超純水系統制備的超純水。
2.2?實驗方法
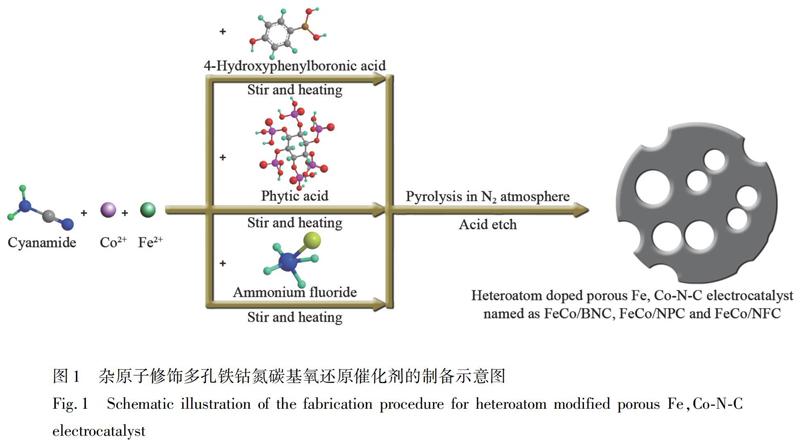
2.2.1?FeCo/NC、FeCo/BNC、FeCo/NPC和FeCo/NFC的合成?首先,將1.06 g FeSO4·7H2O和0.28 g Co(NO3)2·6H2O溶于6 mL水中。在持續攪拌下,加入4 mL單氰胺溶液,在80℃下加熱,得到紫色混合物。將上述混合物在氮氣氛中以4℃/min的升溫速率加熱至800℃,并熱解反應3 h。所得材料在3 mol/L HCl中80℃持續回流8 h,冷卻至室溫,用超純水和無水乙醇洗滌并干燥,制得FeCo/NC。 FeCo/BNC、FeCo/NPC和FeCo/NFC的合成與FeCo/NC的合成步驟基本一致,區別在于Co(NO3)2·6H2O、FeSO4·7H2O與單氰胺混合過程中,引入雜原子前驅體。對于FeCo/BNC的制備,再外加0.01 g對羥基苯硼酸; FeCo/NPC的制備需要外加0.05 mL植酸溶液; FeCo/NFC的制備需要外加0.2 g NH4F。制作流程如圖1所示。
2.2.2?電化學測試及數據計算方法?所有的電化學實驗均在室溫條件下,在氧氣飽和的0.1 mol/L KOH溶液中進行。電化學測試采用三電極系統:玻碳環盤電極作為工作電極, 飽和甘汞電極作為參比電極,鉑電極作為對電極。本研究所提及的電位依據能斯特方程校準:ERHE = ESCE + 0.242+0.059pH。工作電極在修飾前,首先用1.0、0.3和0.05 μm的Al2O3進行拋光,使用乙醇和超純水反復進行超聲清洗, 最后用氮氣吹干電極表面。所用玻碳環盤電極直徑為5.6 mm。工作電極修飾方法如下: 6 mg催化劑、0.2 mL Nafion分散液(5%, m/m)和1.8 mL水混合,超聲處理30 min,以獲得催化劑分散液,將分散液修飾于玻碳電極表面,并在紅外燈下干燥10 min,催化劑修飾量為213.3 μg/cm2。
在每次ORR測試前,所使用的0.1 mol/L KOH溶液需持續通氧15 min,以保證溶液中氧氣飽和。線性掃描伏安(LSV)測試電勢范圍為1.05~0.20 V,掃速5 mV/s,轉速為1600 r/min,環電勢為1.4 V。計時電流法測試電勢為0.6 V。采用LSV實驗數據計算ORR過程中的電子轉移數n和HO2產量,計算公式如下[13]:
其中,IR為環電流, ID為盤電流, N為旋轉環盤電極的收集系數,其值為0.37。
3?結果與討論
3.1??材料組成和結構表征
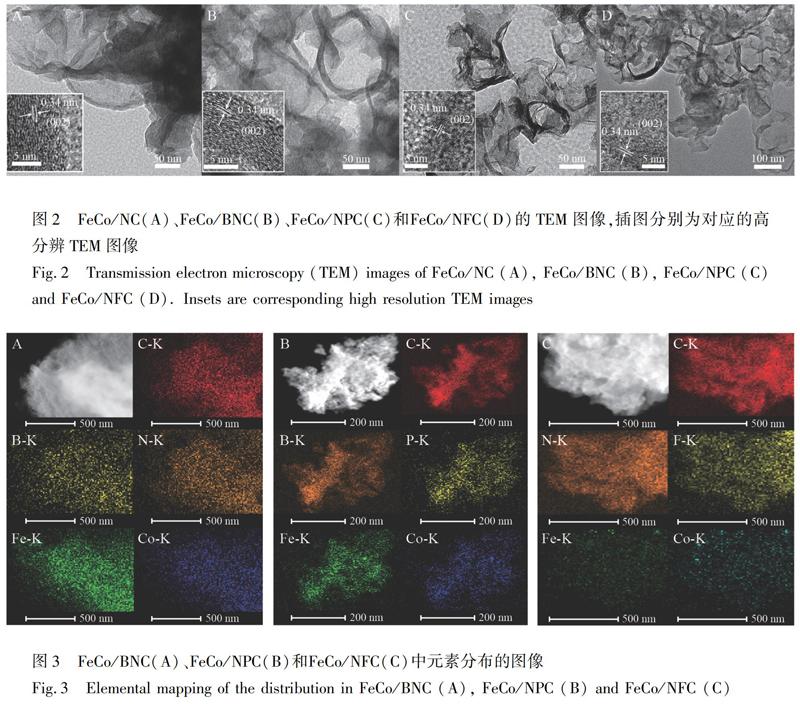
采用透射電鏡(TEM)對所制備材料的結構形貌進行表征。如圖2A所示,FeCo/NC具有多孔碳形貌,這是由于材料內部的金屬粒子被酸刻蝕所致。高分辨TEM圖像(圖2A插圖)表明,多孔碳層具有明顯的晶格,其晶格間距為0.34 nm,對應于石墨碳的(002)晶面[13]。同時,FeCo/BNC、FeCo/NPC和FeCo/NFC的TEM圖像(圖2B~2D)呈現出與FeCo/NC相似的形貌結構,說明雜原子摻雜對材料的結構形貌沒有明顯影響。FeCo/BNC、FeCo/NPC和FeCo/NFC的元素分布圖像(圖3)表明,Fe、Co、N、C以及修飾的雜原子(B、P和F)均勻分散在所制備的材料中; 同時,Fe和Co的分布圖像中并未出現明顯增強的信號,說明采用雜原子進行修飾的過程不會因誘發金屬粒子的留存導致材料多孔性降低。
采用氮氣吸附-脫附測試表征雜原子修飾對材料比表面積和孔徑分布的影響。所制備材料均呈現出IV型等溫曲線(圖4A),明顯的滯后環說明材料中存在大量介孔。FeCo/NC、FeCo/BNC、FeCo/NPC以及FeCo/NFC的比表面積分別為577.7、456.1、850.3和691.4 m2/g。相較于FeCo/NC,FeCo/BNC比表面積的降低主要是由于對羥基苯硼酸在碳化過程中生成部分殘余碳,導致材料多孔性下降(圖4B)[14]。與此相反,植酸可與Fe、Co進行配位,生成比表面積較高的碳化產物,FeCo/NPC的比表面積增加[15]; NH4F不僅能夠進行氟摻雜,熱解過程中產生的氨氣也能刻蝕碳基骨架,改善FeCo/NFC的多孔性[16, 17]。材料的比表面積與其電催化能力在一定程度上呈正相關趨勢,這是由于較高的比表面積能促進催化活性中心的暴露,并加速催化過程中的物質傳遞,從而增加活性中心與催化對象的接觸概率; 同時,比表面積的增加也有利于提升材料的導電性[18]。
采用XPS對所制備材料表面的元素構型進行分析。由圖5A可見,FeCo/NC、FeCo/BNC、FeCo/NPC以及FeCo/NFC具有3種氮構型,即吡啶氮(398.1 eV)、吡咯氮(398.9 eV)和石墨氮(400.8 eV)[19],其中,吡啶氮的存在被認為是由于金屬-氮-碳鍵的形成[20]。 所制備催化劑中XPS不同氮構型占比的對比結果見表1。隨著硼的修飾,FeCo/BNC中吡啶氮和吡咯氮的構型占比明顯增加,這主要是由于BN鍵的形成[21]。對于FeCo/NPC,3種氮構型的比例與FeCo/NC相比沒有明顯的變化,說明使用植酸進行磷摻雜對于氮的構型影響不大。此外,引入氟時,FeCo/NFC中吡啶氮的占比明顯上升,而吡咯氮和石墨氮的占比則呈下降趨勢,此現象歸因于NH4F在熱解過程中釋放的氨氣進行了氮摻雜[22]。圖5B為FeCo/BNC、FeCo/NPC和FeCo/NFC中相應雜原子的XPS圖像,其中,FeCo/BNC中的硼構型可分為B-N(190.7 eV)和B-O(192.6 eV)[23]; FeCo/NPC中的磷構型則分為P-C(132.7 eV)和P-O(133.8 eV)[24]; FeCo/NFC中的氟構型為C-F(686.0 eV)[25]。上述結果說明,采用相應的前驅體可成功地使用雜原子對FeCo/NC進行修飾。圖5C和5D分別為所制備材料的Fe 2p和Co 2p XPS圖譜,并未檢測到明顯的信號,說明材料中的Fe、Co含量低于XPS的檢出限,也再次證明了雜原子的引入不會誘發金屬在材料中的殘留。
3.2??材料的電催化ORR性能
為了探索所制備材料的電催化ORR活性及催化過程,采用旋轉環盤電極(RRDE)在氧氣飽和的0.1 mol/L KOH溶液中進行了LSV測試。如圖 6A中的LSV曲線所示,與商業Pt/C相比,所制備的材料在起始電位方面均有所改善。同時,相較于FeCo/NC,FeCo/BNC具有較差的起始電位和極限擴散電流,這說明比表面積對于提升ORR活性的重要性。對于FeCo/NPC,其極限擴散電流與FeCo/NC相比顯著提升,同樣歸因于催化劑比表面積的提升。此外,FeCo/NFC相比于FeCo/NC在起始電位和極限擴散電流上均有改善,是受益于材料自身比表面積的增加、氮構型的變化以及氟的摻雜。
以RRDE的LSV曲線數據為基礎,依據公式(1)和(2),對所制備材料在催化ORR過程中的電子轉移數以及HO2中間產物產率進行探究(圖 6B)。由表2可知,在所制備的材料中,FeCo/NFC具有最高的電子轉移數和最低的HO2中間產物產率,并與商業Pt/C近似; 同時,相比于FeCo/NPC,FeCo/BNC具有更穩定的電子轉移數。結合相關的XPS數據,修飾硼和氟均可提升吡啶氮的比例,從而促進金屬-氮-碳鍵的形成,有利于ORR催化的四電子選擇性。基于起始電位、極限擴散電流和催化選擇性等指標,選擇FeCo/NFC進行后續測試。
催化劑的穩定性和抗甲醇毒化能力是燃料電池實際應用中的重要考察標準。采用計時電流法對FeCo/NFC和商業Pt/C進行了相關測試。由圖 6C可見,在連續測試20000 s后,FeCo/NFC的電流衰減為7.5%,而Pt/C的衰減值為14.9%。此外,催化劑的抗甲醇毒化能力通過在3 mol/L甲醇條件下進行測試驗證(圖 6D)。在約400 s時將甲醇加入到電解液中,使催化環境中甲醇的濃度達到3 mol/L。FeCo/NFC的電流信號幾乎無影響,然而Pt/C的信號出現大幅衰減。由上述實驗結果可知,FeCo/NFC具有明顯優于Pt/C的電催化ORR穩定性和抗甲醇毒化能力。
4?結 論
采用簡單的碳化-酸刻蝕方法,合成了一系列雜原子修飾的多孔鐵鈷氮碳基材料,并用于高效穩定的ORR催化劑。通過電化學測試,并結合相關組成形貌的表征,研究了雜原子修飾對鐵鈷氮碳基材料ORR催化性能的改進方式,即硼摻雜能夠改進催化的選擇性; 磷摻雜能夠提高催化的活性; 氟摻雜則可以同時提高催化的活性和選擇性。所制備的FeCo/NFC具有可媲美商業Pt/C的催化活性、近四電子的催化選擇性和遠優于Pt/C的催化穩定性和抗甲醇能力,作為Pt/C催化劑的替代品具有較好的應用前景。本研究為研發更高效的摻雜型金屬-氮基ORR催化劑提供了理論基礎。
References
1?Wang D W, Li Q, Han C, Lu Q Q, Xing Z C, Yang X R. Nat. Commun., 2019, 10(1): 3899
2?Huang X X, Shen T, Z T, Qiu H L, Gu X X, Ali Z S, Hou Y L. Adv. Energy Mater., ?2020, ?10(11): 1900375
3?Wu X, Tang C J, Cheng Y, Min X B, Jiang S P, Wang S Y. Chem. Eur. J., ?2020, ?26(18): 3906-3929
4?Wang X Q, Li Z J, Qu Y T, Yuan T W, Wang W Y, Wu Y, Li Y D. Chem, ?2019, ?5(6): 1486-1511
5?Gewirth A A, Varnell J A, DiAscro A M. Chem. Rev., 2018, ?118(5): 2313-2339
6?Huang Y P, Liu W F, Kan S T, Liu P G, Hao R, Hu H, Zhang J, Liu H T, Liu M, Liu K Y. Int. J. Hydrogen Energy, ?2020, ?45(11): 6380-6390
7?HUANG Yan-Ping, YUAN Hong-Yan, ZHANG Jian, YANG Ya-Hui, LIU Hong-Tao. Chinese J. Anal. Chem., ?2017, ?45(9): 1297-1302
黃燕平, 苑紅艷, 張 健, 楊亞輝, 劉洪濤. 分析化學, ?2017, ?45(9): 1297-1302
8?He Y H, Tan Q, Lu L L, Sokolowski J, Wu G. Electrochem. Energy Rev., ?2019, ?2(2): 231-251
9?Sultan S, Tiwari J N, Jang J, Harzandi A M, Salehnia F, Yoo S J, Kim K S. Adv. Energy Mater., ?2018, ?8(25): 1801002
10?Huang Y P, Liu W F, Kan S T, Liu P G, Liu H T, Liu K Y. ChemistrySelect, ?2019, ?4(36): 10863-10867
11?Lv Q, Si W Y, He J J, Sun L, Zhang C F, Wang N, Yang Z, Li X D, Wang X, Deng W Q, Long Y Z, Huang C S, Li Y L. Nat. Commun., ?2018, ?9(1): 3376
12?Shah M S A S, Lee J, Rauf A, Park J H, Lim B, Yoo P J. Nanoscale, ?2018, ?10(41): 19498-19508
13?Han C, Li Q, Wang D W, Lu Q Q, Xing Z C, Yang X R. Small, ?2018, ?14(17): 1703642
14?Bo X J, Guo L P. PCCP, ?2013, ?15(7): 2459-2465
15?Ma Y M, Gan L, Li D, Gao Y Y, Yang X X, Wang K, Lu S Y, Wu H, Ding S J, Xiao C H. J. Power Sources, ?2019, ?441: 227177
16?Sun X J, Song P, Chen T, Liu J, Xu W L. Chem. Commun., ?2013, ?49(87): 10296-10298
17?Wang X Q, Lee J S, Zhu Q, Liu J, Wang Y, Dai S. Chem. Mater., ?2010, ?22(7): 2178-2180
18?Wang Y, Guo N N, Zhu L K, Pan Y, Wang R W, Zhang Z T, Qiu S L. Chem. Commun., ?2018, ?54(92): 12974-12977
19?Ren X H, Guo H H, Ma X X, Hou G M, Chen L, Xu X Y, Chen Q, Feng J K, Si P C, Zhang L, Ci L J. Appl. Surf. Sci., ?2018, ?457: 780-788
20?Sun T T, Zhang S L, Xu L B, Wang D S, Li Y D. Chem. Commun., ?2018, ?54(85): 12101-12104
21?Han C, Bo X J, Zhang Y F, Li M, Wang A X, Guo L P. Chem. Commun., ?2015, ?51(81): 15015-15018
22?Geng D S, Chen Y G, Chen Y G, Li Y L, Li R Y, Sun X L, Ye S Y, Knights S. Energy ?Environ. Sci., ?2011, ?4(3): 760-764
23?Fei H L, Ye R Q, Ye G L, Gong Y J, Peng Z W, Fan X J, Samuel E L G, Ajayan P M, Tour J M. ACS Nano, ?2014, ?8(10): 10837-10843
24?Ling W, Wang Z A, Ma Q, Deng Q, Tang J F, Deng L, Zhu L H, Wu X W, Yue J P, Guo Y G. Chem. Commun., ?2019, ?55(77): 11515-11518
25?Bhowmick S, Khan M Z U, Banerji A, Lukitsch M J, Alpas A T. Appl. Surf. Sci., ?2018, ?450: 274-283
