
先天性膽汁酸合成障礙
2020-10-28 01:32:46李娟
健康必讀·下旬刊 2020年10期
李娟
【摘 要】目的:探討先天性膽汁酸合成障礙患兒的臨床特點、診斷、治療及預后。方法:回顧性分析1例先天性膽汁酸合成障礙患兒的臨床特點、治療、預后,并對家族進行基因分析。結果:該病患兒有膽汁淤積表現,但γ-GTT下降或正常,總膽汁酸下降或正常,基因檢測可明確診斷,在治療上,早期予初級膽汁酸替代治療可明顯改善病情,若出現肝硬化,需肝移植治療。結論:盡快完善基因檢查,及早診斷及治療,改善患兒的預后及生存治療,同時為家系遺傳咨詢及產前診斷提供依據。
【關鍵詞】膽汁淤積;先天性;基因檢測
【中圖分類號】R541【文獻標識碼】B【文章編號】1672-3783(2020)10-30--01
先天性膽汁酸合成障礙(congenitalbileacidsynthesisdefect,CBAS)是一種罕見遺傳性疾病,多為常染色體隱性遺傳,是由于合成兩種主要膽汁酸所必需的酶存在遺傳缺陷,而引起先天性膽汁酸合成障礙,約占兒童膽汁淤積性肝病的1%-2%。膽汁酸是在肝細胞內以膽固醇為原料,經過一系列的酶促反應、形成;膽汁酸合成過程中需要至少14種酶參與,任何一個酶的缺乏都將導致正常膽汁酸生成障礙,從而導致一系列疾病的發生。其中,3β-羥基-△5-C27-類固醇脫氫酶缺陷是先天性膽汁酸合成障礙中報道最多的一類酶缺陷疾病,為先天性膽汁酸合成障礙1型(CBAS1),是由16號染色體HSD3B7基因突變引起,為常染色體隱性遺傳。我對1例先天性膽汁酸合成障礙患兒臨床表現、基因進行分析,旨在進一步了解和掌握改變的臨床特殊、早期診斷及治療。
1 臨床資料
患兒,男,6月7天,籍貫為c云南曲靖市,因“發現皮膚黃染1月余”2019年6月1日入住昆明市兒童消化內科。
現病史:1個月余前患兒皮膚、鞏膜出現黃染,無發熱,無嗜睡、抽搐,無尿色加深,大便無變白。無咳嗽、吐沫,有吐奶,無腹瀉、腹脹。大便為淡黃色糊狀便,小便顏色清亮,體重無增長。
家族史:患兒G4P4,足月順產,出生體重3.6公斤,否認黃疸病史,否認家族內類似病史。
體格檢查:體重5.9kg,一般情況及精神欠佳,神清,輕度貧血貌,營養不良貌,全身皮膚重度黃染,鞏膜黃染,無皮疹、瘀斑、淤點,前囟平軟,口周無發紺,咽無充血,雙肺呼吸音粗,可聞及大量粗濕羅音,心律齊,心音有力,腹脹,肝右肋下6cm,劍突下4.0cm,質硬,邊銳,脾肋下4.0cm,質硬,邊銳,未捫及包塊,腸鳴音正常。雙下肢不腫,肢端暖,神經系統檢查無異常。
輔助檢查:2019年05月30日我院血常規:WBC8.91ⅹ10^9/L,N30.10%,L55.60%,RBC3.71ⅹ10^12/L,Hb83.0g/L,PLT307.0ⅹ10^9/L,CRP2.05mg/L,網織紅細胞百分比1.57%。5.30肝功:ALT71U/L,AST385U/L,總膽紅素150.0umol/L,間接膽紅素82.0umol/L,直接膽紅素67.3umol/L,γ-GGT:21U/L,總膽汁酸16.5umol/L。腎功、心肌酶、葡萄糖、電解質:正常。2019-06-02凝血7項:凝血酶原時間(stago)16.5秒,纖維蛋白原(stago)1.38g/L,血漿抗凝血酶III活性測定(stago)44%,活化部分凝血活酶時間(stago)52.3秒。2019-06-02血脂:總膽固醇3.00mmol/L,載脂蛋白A0.74g/L,甘油三酯0.97mmol/L。甲胎蛋白:60500ng/ml。血氨:正常。乙肝+甲肝+丙肝抗體:陰性。2019-06-0225-羥維生素D(VITD-T):34.08nmol/L;2019-06-05血清維生素測定:維生素E13.05umol/L,維生素A0.35umol/L。
血尿遺傳代謝篩查:未見明顯異常。
影像學檢查:2019-06-05胰膽管水成像(MRCP):膽囊小,壁增厚,膽囊窩積液;肝臟右葉體積稍大,肝裂稍寬,余膽道、脾臟、胰腺、雙腎未見異常;少量腹腔積液;掃描層面雙肺下葉炎變。
診斷、治療及隨訪:患兒入院后予熊去氧膽酸口服治療,療效不佳,評估患兒已有肝硬化,家長不愿行肝移植術,出院后半年死亡。
2基因分析
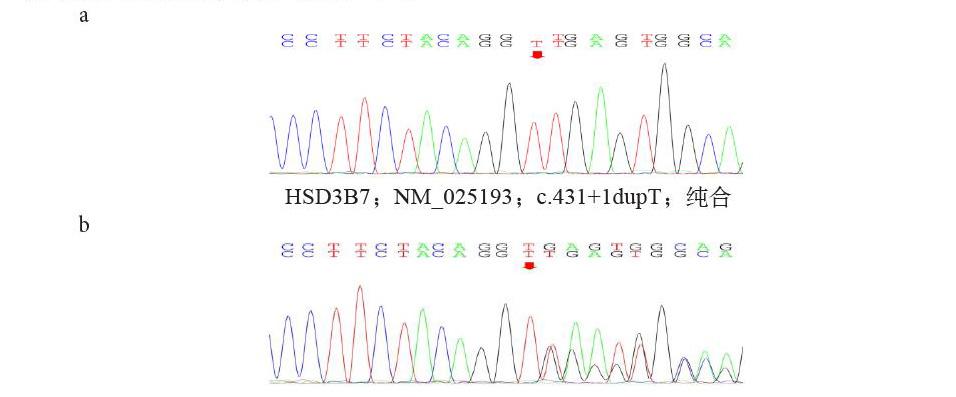
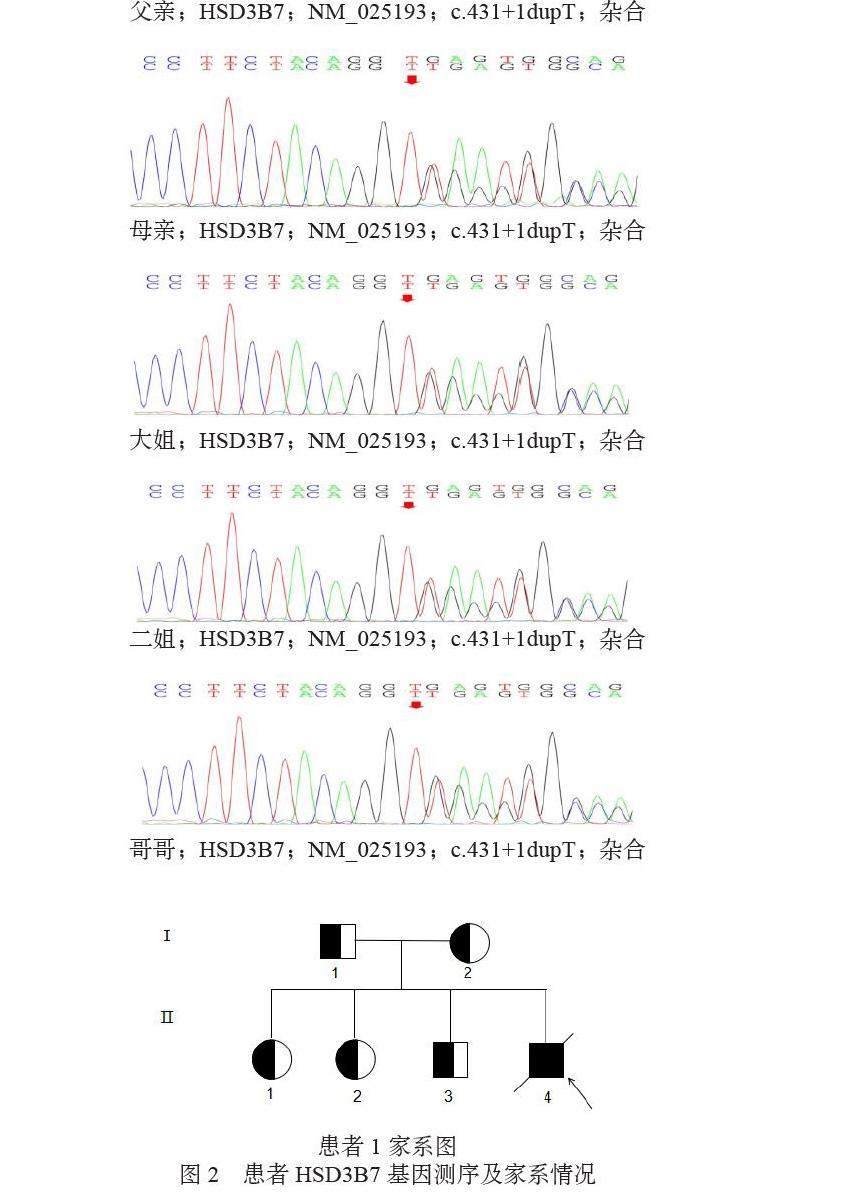
患者經膽汁淤積癥檢測項目,發現HSD3B7基因存在一剪切變異,為c.431+1dupT,即基因的編碼序列第431位的堿基后的1個堿基T存在重復,經Sanger測序驗證證實(見圖2a),患兒上述突變為純合變異,同時對其家系進行了驗證(見圖2b),父親母親均為突變攜帶者,并對家系情況進行了分析(見圖2c)。
3 討論
先天性膽汁酸合成障礙呈常染色體隱性遺傳,致病基因攜帶者不會有臨床表現,當夫妻雙方均為攜帶者時有25%的幾率將致病基因遺傳給后代并致病,故當均為攜帶者的夫妻在生育時可通過產前診斷或植入前遺傳學診斷預防再生育患兒。
先天性膽汁酸合成障礙應盡早使用初級膽汁酸(膽酸CA或鵝脫氧膽酸CFCA),一方面可負反饋抑制7α-羥化酶活性,從而抑制7α-羥化膽固醇異常異常代謝產物,降低對肝臟毒性損傷;另一方面刺激膽汁液形成,促進肝臟清除內源性膽汁酸、膽紅素和毒性物質的排出。熊去氧膽酸不能抑制7α-羥化酶活性,但對先天性膽汁酸合成障礙1型患者使用熊去氧膽酸,肝損傷、膽汁淤積可好轉,提示熊去氧膽酸對先天性膽汁酸合成障礙1型有一定療效。先天性膽汁酸合成障礙2型使用熊去氧膽酸后,肝損傷、膽汁淤積無改善,無明顯療效,使用鵝脫氧膽酸可改善。故需早期使用初級膽汁酸,但若患兒病情不能控制或有肝硬化,口服初級膽汁酸已無法緩解病情,需行肝移植術治療。
綜上所述,嬰幼兒出現膽汁淤積性肝病,血清總膽汁酸下降或正常,γ-GTT下降或正常,需考慮膽汁酸合成障礙,盡快完善基因檢查,及早診斷及治療,改善患兒的預后及生存治療,同時為家系遺傳咨詢及產前診斷提供依據。
