固相萃取—高效液相色譜—串聯(lián)質(zhì)譜法測(cè)定動(dòng)物源食品中萬(wàn)古霉素類抗生素殘留量
2020-10-19 00:32:34范小龍豐彭青枝周陶鴻
食品與機(jī)械 2020年9期
林 津 范小龍 江 豐彭青枝 周陶鴻
(1. 湖北省食品質(zhì)量安全監(jiān)督檢驗(yàn)研究院,湖北 武漢 430075;2. 湖北省食品質(zhì)量安全檢測(cè)工程技術(shù)研究中心,湖北 武漢 430075)
萬(wàn)古霉素類抗生素是由鏈霉菌產(chǎn)生的一類具有復(fù)雜結(jié)構(gòu)的糖肽類抗生素,其中典型的代表物有萬(wàn)古霉素和去甲萬(wàn)古霉素[1]。該類抗生素通過抑制肽聚糖的生物合成,可對(duì)革蘭氏陽(yáng)性菌起到強(qiáng)力的滅殺作用,由于其藥效顯著,常常使用于其他抗生素對(duì)病菌無效時(shí),即所謂的臨床“最后一線”抗生素[2]。雖然中國(guó)農(nóng)業(yè)農(nóng)村部公布的食品動(dòng)物禁用的獸藥及其他化合物清單中萬(wàn)古霉素赫然在列,但由于獸藥生產(chǎn)經(jīng)營(yíng)秩序不夠規(guī)范,獸藥濫用現(xiàn)象仍然較為嚴(yán)重,不法企業(yè)或養(yǎng)殖戶可能將其用于動(dòng)物養(yǎng)殖,減少動(dòng)物疫病的發(fā)生,以擴(kuò)大養(yǎng)殖利潤(rùn)[3-4]。可靠的殘留檢測(cè)方法是進(jìn)行農(nóng)產(chǎn)品抽檢和市場(chǎng)監(jiān)管的基礎(chǔ)和前提。因此,建立動(dòng)物源食品中萬(wàn)古霉素類抗生素殘留量檢測(cè)方法,加強(qiáng)相關(guān)食品的監(jiān)控是非常必要的。
目前現(xiàn)行有效的針對(duì)萬(wàn)古霉素類抗生素的檢測(cè)方法標(biāo)準(zhǔn)只有農(nóng)業(yè)部1862號(hào)公告-3-2012《飼料中萬(wàn)古霉素的測(cè)定 液相色譜—串聯(lián)質(zhì)譜法》和吉林地方標(biāo)準(zhǔn)DB22/T 1825—2013《豬肉中萬(wàn)古霉素殘留量的測(cè)定 液相色譜—質(zhì)譜/質(zhì)譜法》,標(biāo)準(zhǔn)適用性較窄,難以廣泛應(yīng)用,不利于中國(guó)動(dòng)物性食品安全監(jiān)管。而關(guān)于萬(wàn)古霉素類化合物的分析方法已見報(bào)道的有:液相色譜法、液相色譜—串聯(lián)質(zhì)譜法、酶放大免疫法、毛細(xì)管電泳化學(xué)發(fā)光法、熒光偏振免疫法、近紅外光譜法、微生物法[5-7]等,分析對(duì)象主要是集中在人體樣本如血清、血漿和尿液[8-10]等。而針對(duì)動(dòng)物源食品的分析研究并不多,且基質(zhì)較單一,基本只有單獨(dú)檢測(cè)豬肉、牛乳和魚蝦等一種基質(zhì),且目標(biāo)物主要集中在萬(wàn)古霉素這一種物質(zhì)上,導(dǎo)致此類抗生素檢測(cè)覆蓋不全,不利于檢測(cè)資源的優(yōu)化配置和監(jiān)管效能的提高[1-5]。所以對(duì)于脂肪含量較高、基質(zhì)成分復(fù)雜的動(dòng)物源食品中萬(wàn)古霉素類抗生素同時(shí)測(cè)定的研究還較少,且提供的方法在實(shí)際操作過程中相對(duì)復(fù)雜、穩(wěn)定性差[6-7]。研究擬采用固相萃取凈化—高效液相—串聯(lián)質(zhì)譜聯(lián)用的分析方法,對(duì)日常食用的動(dòng)物源性食品基質(zhì)中萬(wàn)古霉素類抗生素的殘留進(jìn)行檢測(cè),以期為獸藥殘留監(jiān)管提供技術(shù)支撐。
1 材料與方法
1.1 材料與試劑
豬肉、雞肉、鴨肉、豬肝:武漢市售;
萬(wàn)古霉素和去甲萬(wàn)古霉素標(biāo)準(zhǔn)品:純度均>98%;美國(guó)MCE公司;
雙去氯萬(wàn)古霉素標(biāo)準(zhǔn)品:純度>75%;加拿大Toronto公司;
乙腈、甲酸、甲醇:色譜純,德國(guó)Merck公司;
氨水、鹽酸、正己烷:分析純,國(guó)藥集團(tuán)化學(xué)試劑有限公司;
OasisMCX固相萃取柱:美國(guó)Waters公司;
試驗(yàn)用水:一級(jí)水,美國(guó)Mill-pore-Q超純水儀制得。
1.2 儀器與設(shè)備
高效液相色譜儀:Ultimate 3000型,美國(guó)Thermo公司;
串聯(lián)四極桿質(zhì)譜儀:AB sciex 4500型,美國(guó)Applied Biosystems公司;
電子天平:ME2002E型和XS204型,瑞士Mettler-Toledo公司;
超聲清洗器:Elmasonic P60H型,德國(guó)Elma公司;
臺(tái)式冷凍離心機(jī):Allegra X-15R型,美國(guó)Beckman公司;
水浴氮吹儀:N-EVAP24型,美國(guó)Organomation公司;
數(shù)顯漩渦混合器:EOFO-9456型,美國(guó)Taloys公司。
風(fēng)電機(jī)組的聯(lián)軸器是整機(jī)中故障率比較高的部件,而且聯(lián)軸器發(fā)生失效后,對(duì)機(jī)組的安全、穩(wěn)定運(yùn)行有很大的影響。失效型式和預(yù)防措施,可為其他風(fēng)電機(jī)組聯(lián)軸器的設(shè)計(jì)、測(cè)試驗(yàn)證及降低失效率提供了參考。
1.3 標(biāo)準(zhǔn)溶液配制
準(zhǔn)確稱取25 mg萬(wàn)古霉素標(biāo)準(zhǔn)品和去甲萬(wàn)古霉素標(biāo)準(zhǔn)品,于不同的25 mL容量瓶中,用甲醇充分溶解,再定容至刻度線,搖勻,配制成1 mg/mL的標(biāo)準(zhǔn)儲(chǔ)備液。準(zhǔn)確稱取雙去氯萬(wàn)古霉素標(biāo)準(zhǔn)品10 mg(精確至0.000 1 g),于10 mL容量瓶中,用甲醇充分溶解,再定容至刻度線,搖勻,配制成1 mg/mL的內(nèi)標(biāo)儲(chǔ)備液。以上儲(chǔ)備液均置于4 ℃保存,有效期1個(gè)月。
用0.1%甲酸水溶液稀釋,配制成濃度為1 μg/mL的萬(wàn)古霉素和去甲萬(wàn)古霉素混合標(biāo)準(zhǔn)中間液以及1 μg/mL的雙去氯萬(wàn)古霉素內(nèi)標(biāo)中間液,4 ℃條件下可保存1個(gè)月。臨用前,分別準(zhǔn)確移取適量的1 μg/mL的萬(wàn)古霉素和去甲萬(wàn)古霉素混合標(biāo)準(zhǔn)中間液以及雙去氯萬(wàn)古霉素內(nèi)標(biāo)中間液,用0.1%甲酸水溶液進(jìn)行稀釋,制成內(nèi)標(biāo)濃度為100 μg/L的標(biāo)準(zhǔn)系列工作溶液(20~200 μg/L)。
1.4 樣品預(yù)處理
1.4.1 樣品提取 稱取5.00 g經(jīng)組織搗碎機(jī)絞碎并充分均質(zhì)后的樣品于50 mL離心管中,加入100 μL濃度為1 μg/mL 的雙去氯萬(wàn)古霉素內(nèi)標(biāo)中間液,加入10 mL 0.1% 甲酸—乙腈提取液(體積比70∶30),加蓋渦旋混勻1 min,并超聲提取15 min。然后在4 ℃、9 500 r/min離心10 min,將上清轉(zhuǎn)移至50 mL離心管中。殘?jiān)?0 mL 上述提取液重復(fù)提取一次,合并兩次上清,加入5 mL 乙腈飽和正己烷,渦旋混勻1 min,4 ℃、9 500 r/min 離心10 min,取下層清液,待凈化。
1.4.2 樣品凈化 Oasis MCX固相萃取柱使用前依次用3 mL甲醇、3 mL水、3 mL 0.1 mol/L鹽酸溶液活化,保持柱體濕潤(rùn)。取全部待凈化液加入活化后的固相萃取柱,上樣后依次用3 mL 0.1 mol/L鹽酸溶液,3 mL甲醇淋洗,棄去全部淋洗液。最后用6 mL流速穩(wěn)定1 mL/min 內(nèi)的5%氨水—甲醇溶液洗脫,并收集洗脫液,置于40 ℃水浴氮?dú)獯蹈桑? mL 0.1%甲酸水溶液—乙腈溶液(體積比90∶10)復(fù)溶,過微孔濾膜(0.22 μm)后,上HPLC-MS/MS測(cè)定。
1.5 色譜條件
色譜柱選用Waters ACQUITY UPLC BEH C18柱,規(guī)格型號(hào)為2.1 mm×100 mm,2.2 μm;流速0.3 mL/min;柱溫35 ℃;進(jìn)樣量5 μL;HPLC梯度洗脫條件見表1。
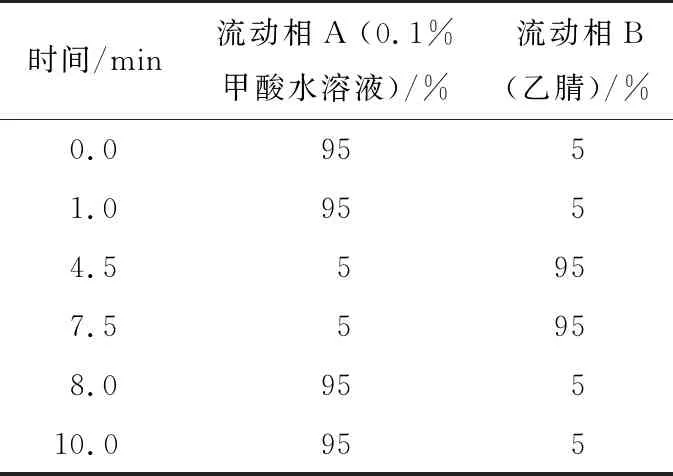
表1 HPLC梯度洗脫程序
1.6 質(zhì)譜條件
離子源類型:ESI源;掃描方式:MRM正離子模式掃描;離子源溫度(TEM):550 ℃;霧化氣(Gas1):34 MPa;輔助氣(Gas2):38 MPa;氣簾氣(Gurtain Gas):25 MPa;電噴霧電壓:4 500 V;通過儀器優(yōu)化后的萬(wàn)古霉素、去甲萬(wàn)古霉素及雙去氯萬(wàn)古霉素的質(zhì)譜參數(shù)見表2。
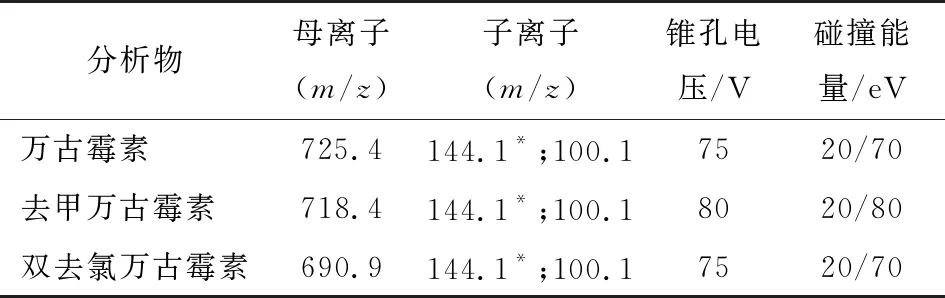
表2 目標(biāo)物和內(nèi)標(biāo)的質(zhì)譜參數(shù)?
2 結(jié)果與分析
2.1 質(zhì)譜條件的確立
萬(wàn)古霉素、去甲萬(wàn)古霉素和雙去氯萬(wàn)古霉素的電噴霧離子化效果較好,故采用電噴霧離子化源進(jìn)行分析。根據(jù)物質(zhì)化學(xué)性質(zhì)和結(jié)構(gòu),通常選擇豐度最高的離子對(duì)為母離子[11],同時(shí)在正負(fù)模式下對(duì)比母離子響應(yīng)強(qiáng)度,最終選擇在正離子模式下進(jìn)行分析。將高濃度的目標(biāo)物和內(nèi)標(biāo)溶液在正離子模式下通過一級(jí)掃描找到母離子,對(duì)選定的母離子加優(yōu)化好的碰撞能擊碎,同時(shí)進(jìn)行二級(jí)掃描找到對(duì)應(yīng)的碎片離子[12]。萬(wàn)古霉素類抗生素是由多個(gè)氨基酸和糖縮合而成的糖肽類大分子化合物,進(jìn)入質(zhì)譜后很容易與H+結(jié)合,產(chǎn)生雙電荷或三電荷離子[13]。經(jīng)試驗(yàn)?zāi)繕?biāo)物和內(nèi)標(biāo)均選擇豐度最大的雙電荷m/z725.4,718.4,690.9作為母離子,定性離子取干擾較小、豐度較高的兩個(gè)質(zhì)荷比分別為144.1和100.1碎片離子[14],且豐度最高m/z144.1作為定量離子。在選擇好的特征碎片離子的基礎(chǔ)上,對(duì)產(chǎn)生碎片離子的碰撞能量、錐孔電壓等其他質(zhì)譜參數(shù)進(jìn)一步優(yōu)化來提高分析方法的靈敏度,進(jìn)一步優(yōu)化后最終確立的質(zhì)譜重要參數(shù)及相應(yīng)的質(zhì)譜條件見1.6。
2.2 色譜條件的確立
2.3 前處理方法的優(yōu)化
2.3.1 提取試劑的選擇 在陰性豬肉基質(zhì)中添加了100 μg/kg 目標(biāo)物,考察了0.1%甲酸溶液和0.1%甲酸—乙腈體積比分別為90∶10,80∶20,70∶30,60∶40的溶液提取效果。單純用0.1%甲酸溶液提取動(dòng)物源樣品時(shí),由于基質(zhì)中的蛋白沒有得到有效沉淀,提取液較渾濁,必須提高乙腈比例沉淀蛋白。隨著乙腈相比例的提高,提取液逐漸澄清,但乙腈相比例超過30%時(shí)回收率無法進(jìn)一步提升,且基質(zhì)效應(yīng)明顯,結(jié)果如圖2所示,最終選擇0.1%甲酸—乙腈(體積比70∶30)溶液作為提取液。
2.3.2 凈化條件的優(yōu)化 動(dòng)物源食品中難免有較多的脂類雜質(zhì),經(jīng)0.1%甲酸—乙腈(體積比70∶30)溶液提取后,仍不足以去除脂類雜質(zhì)的干擾,于是增加了正己烷除脂步驟。由于目標(biāo)物微溶于乙腈,而乙腈與正己烷還存在少量互溶,若要避免萬(wàn)古霉素類抗生素在萃取時(shí)的損失,使用乙腈飽和的正己烷除脂效果更好[17]。加入5 mL的乙腈飽和正己烷,渦旋混勻1 min,可以看到正己烷層明顯有油脂析出,說明除脂效好。
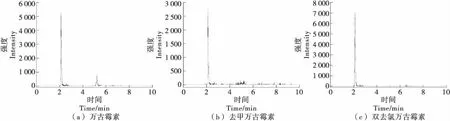
圖1 添加目標(biāo)物樣本的選擇離子流色譜圖Figure 1 Selected ion chromatogram of samples for adding objects
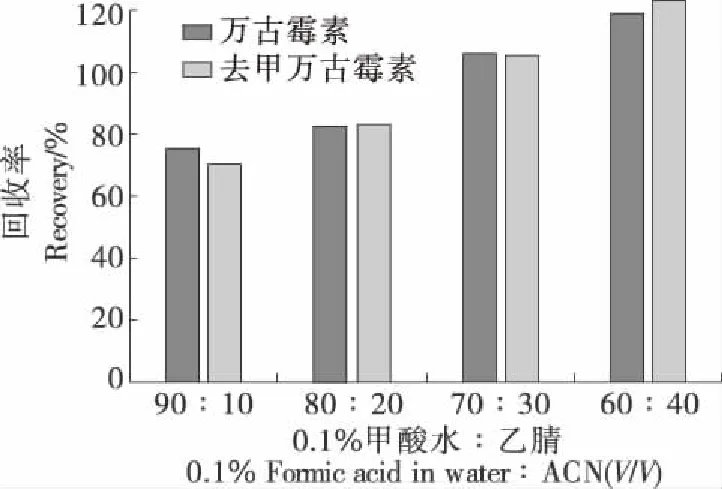
圖2 不同配比的0.1%甲酸水溶液—乙腈提取液效果比較Figure 2 Extraction efficiency with different solutions
試驗(yàn)檢測(cè)對(duì)象為動(dòng)物源樣品,基質(zhì)較復(fù)雜,若直接提取后上機(jī),不進(jìn)行進(jìn)一步的凈化處理,提取液中一并提取的其他雜質(zhì)必定會(huì)對(duì)檢測(cè)帶來干擾和影響,從而影響最終結(jié)果的準(zhǔn)確性,因此必需要增加凈化處理步驟[18]。由于含有堿性的伯氨基團(tuán),目標(biāo)物偏弱堿性[5],將市面上主流廠家的固相萃取柱,如Waters、Agela、Thermo、Agilent的陽(yáng)離子交換柱MCX柱、C18柱和HLB柱等進(jìn)行對(duì)比。配制濃度為60 μg/L的目標(biāo)物混合標(biāo)液,采用標(biāo)準(zhǔn)溶液直接過固相萃取柱凈化的方式考察不同固相萃取柱的影響。結(jié)果表明,陽(yáng)離子交換柱(3 mL)凈化,目標(biāo)物回收率為90%~95%;HLB柱(3 mL)凈化,萬(wàn)古霉素和去甲萬(wàn)古霉素的回收率為50%~70%;用C18柱(3 mL)凈化時(shí),萬(wàn)古霉素和去甲萬(wàn)古霉素的回收率為30%~40%。基于回收率的對(duì)比情況,故用陽(yáng)離子交換柱MCX柱來凈化萬(wàn)古霉素、去甲萬(wàn)古霉素。
2.4 基質(zhì)效應(yīng)的消除
基質(zhì)效應(yīng)(ME)是影響定量分析結(jié)果準(zhǔn)確性的重要因素,是通過陰性基質(zhì)匹配標(biāo)線與純?nèi)軇┡渲茦?biāo)曲二者的斜率之比來進(jìn)行評(píng)價(jià)的[11]。分別配制濃度范圍為20~200 μg/L的陰性基質(zhì)匹配的標(biāo)準(zhǔn)溶液和純?nèi)軇?biāo)準(zhǔn)溶液,計(jì)算兩者斜率比值。針對(duì)動(dòng)物源食品基質(zhì),萬(wàn)古霉素和去甲萬(wàn)古霉素的ME均在70%左右,說明存在一定程度的離子抑制效應(yīng)。為消除基質(zhì)效應(yīng)使定量更加精準(zhǔn),既可采取基質(zhì)匹配外標(biāo)曲線校正法,也能采用內(nèi)標(biāo)法定量。選擇內(nèi)標(biāo)物時(shí),不僅要求內(nèi)標(biāo)物與目標(biāo)物的物理化學(xué)性質(zhì)相似,同時(shí)不與被測(cè)定樣品發(fā)生化學(xué)反應(yīng),且應(yīng)與目標(biāo)物的出峰時(shí)間相近等[19]。試驗(yàn)選用的內(nèi)標(biāo)物——雙去氯萬(wàn)古霉素的結(jié)構(gòu)只比萬(wàn)古霉素分子結(jié)構(gòu)的少2個(gè)氯,所以其結(jié)構(gòu)和性質(zhì)與萬(wàn)古霉素基本無差異,可以滿足內(nèi)標(biāo)物的要求[20]。目前使用該內(nèi)標(biāo)物進(jìn)行萬(wàn)古霉素和去甲萬(wàn)古霉素檢測(cè)的研究較少,主要分布在魚蝦基質(zhì)中。
2.5 線性范圍、檢出限(LOD)與定量限(LOQ)
配制濃度依次為20,40,80,120,160,200 μg/L的混合系列標(biāo)準(zhǔn)工作溶液,在上述優(yōu)化后的液相條件、質(zhì)譜參數(shù)及最終確立的分析方法下進(jìn)行測(cè)定。以目標(biāo)物的濃度(μg/L)為x,萬(wàn)古霉素或去甲萬(wàn)古霉素與內(nèi)標(biāo)雙去氯萬(wàn)古霉素的峰面積比值為y,進(jìn)行線性曲線擬合。如表2所示,在20~200 μg/L的線性范圍內(nèi),目標(biāo)物線性關(guān)系良好,相關(guān)系數(shù)R2均可達(dá)到0.997以上。將陰性基質(zhì)加標(biāo)樣按優(yōu)化后的前處理方法進(jìn)行處理,同時(shí)按最終確立的分析方法進(jìn)行檢測(cè),以目標(biāo)物信號(hào)噪聲比值(S/N)為3時(shí)的加標(biāo)水平作為L(zhǎng)OD,以S/N為10時(shí)的加標(biāo)水平作為L(zhǎng)OQ,方法檢出限可低至1.0 μg/kg,定量限可達(dá)到3.0 μg/kg。

表3 目標(biāo)物的線性范圍、回歸方程、相關(guān)系數(shù)、檢出限和定量限
2.6 方法的回收率與精密度
分別取豬肉、雞肉、鴨肉、豬肝4種基質(zhì),基質(zhì)中均不含試驗(yàn)方法所檢測(cè)的目標(biāo)物,按照20,50,200 μg/kg 3個(gè)濃度水平分別添加標(biāo)樣,每個(gè)水平做6個(gè)平行樣,按上述方法進(jìn)行分析,計(jì)算回收率和精密度,具體數(shù)據(jù)見表4。

表4 試驗(yàn)方法的回收率與精密度
結(jié)果表明,目標(biāo)物的平均加標(biāo)回收率為88.7%~115.1%,相對(duì)標(biāo)準(zhǔn)偏差(RSD)為3.1%~9.4%(n=6)。按照GB/T 27404—2008標(biāo)準(zhǔn),在被測(cè)組分含量小于0.1 mg/kg時(shí)回收率的要求為60%~120%,被測(cè)組分含量在0.1~1.0 mg/kg 時(shí)回收率的要求為80%~110%。因此,試驗(yàn)方法回收率和精密度均滿足要求。
3 結(jié)論
試驗(yàn)建立了一種動(dòng)物源食品中萬(wàn)古霉素類抗生素殘留量的高效液相色譜—串聯(lián)質(zhì)譜檢測(cè)分析方法,通過選擇提取效率更高的提取液,除脂效果更好的萃取液,陽(yáng)離子固相萃取柱凈化的一系列前處理步驟更進(jìn)一步的減少了雜質(zhì)干擾,且對(duì)比考察了更優(yōu)的色譜及質(zhì)譜條件,同時(shí)使用雙去氯萬(wàn)古霉素作為內(nèi)標(biāo)物進(jìn)行定量分析,有效提高了檢測(cè)的靈敏度和準(zhǔn)確度,并進(jìn)一步降低了檢出限。在20~200 μg/kg添加水平范圍內(nèi)的平均回收率為88.7%~115.1%,相對(duì)標(biāo)準(zhǔn)偏差為3.1%~9.4%(n=6),方法檢出限低至1.0 μg/kg,定量限達(dá)到3.0 μg/kg。該方法回收率高,檢出限低,分析時(shí)間短,適用于批量的動(dòng)物源食品中萬(wàn)古霉素類抗生素殘留量的同時(shí)檢測(cè)。試驗(yàn)建立的方法在已報(bào)道的豬肉制品、水產(chǎn)品等基質(zhì)基礎(chǔ)上又有了進(jìn)一步的擴(kuò)充和驗(yàn)證,同時(shí)又由于合適的內(nèi)標(biāo)的引入比基質(zhì)匹配的方法在基質(zhì)消除這一方面更簡(jiǎn)便快捷,使得試驗(yàn)方法基質(zhì)覆蓋更全、適用性更廣、實(shí)用性更強(qiáng)。以復(fù)雜基質(zhì)樣品為分析對(duì)象時(shí),固相萃取—高效液相色譜—串聯(lián)質(zhì)譜的分析方法的高分離性、高選擇性、高靈敏度以及能獲取目標(biāo)物結(jié)構(gòu)信息[7]等一系列優(yōu)點(diǎn),能夠滿足痕量分析檢測(cè)的要求,將在動(dòng)物源食品抗生素殘留量抽檢工作方面發(fā)揮越來越重要的作用。
