快速水蒸氣蒸餾-流動注射分析儀聯用測定食品中氰化物
2020-10-18 08:31:18馬興張密密肖亞兵趙友全
食品研究與開發 2020年19期
馬興,張密密,肖亞兵,趙友全
(1.天津海關動植物與食品檢測中心,天津300461;2.天津大學精密儀器與光電子工程學院,天津300072)
氰化物屬劇毒物質,由氰化物引發的食物中毒、環境污染[1-4]對突發公共安全事件管控和處置提出了嚴峻的挑戰。食品中氰化物主要來源于植物本身所含有的氰甙類物質,文獻報道顯示目前已知含有氰甙的植物有至少2 000種,包含木薯、豆類、亞麻籽、甘藍、苦杏仁和食用菌等[5]。當氰甙進入人體內后,在酸和酶的作用下會釋放出游離氰,氰離子具有很強的金屬配位能力,能夠抑制細胞內多種含金屬離子的酶系統,致使如細胞色素氧化酶、乳酸脫氫酶和過氧化氫酶等酶失活,從而導致中毒現象[6-8]。輕度氰化物中毒有明顯頭痛、頭暈、惡心、嘔吐、乏力等癥狀,嚴重者表現為昏迷、肌肉痙攣、嚴重代謝性酸中毒甚至猝死。
目前,測定食品中氰化物含量的國家標準GB 5009.36-2016《食品安全國家標準食品中氰化物的測定》規定了氫氰酸與苦味酸鈉的定性顯色方法,分光光度法和氣相色譜法兩種定量檢測方法[9]。其中分光光度法需要先通過水蒸氣蒸餾將氰化物以氫氰酸的形式蒸出,而傳統水蒸氣蒸餾時間長、操作復雜、重現性較差,導致該方法檢測效率較低,對檢測人員的操作技能和檢測經驗要求較高。氣相色譜法定量爭議較大,目前標準所述操作同樣可以用來檢測硫氰根離子[10-12],因而實際檢測過程中難以消除硫氰酸鹽的干擾。
流動注射分析法具有自動化高,進樣量小,試劑消耗量小,環保性高,檢測量大,穩定性強等優點,最初只用于水質氰化物[13]、揮發酚[14]等項目的檢測。近年來,流動注射法在食品和飲料中硫酸鹽[15]、牛奶中青霉素G[16]、木薯中氰化物[17]、海水中痕量錳[18]、蒸餾酒中氰化物[19]、葡萄酒中總糖[20]、生鮮乳中硫氰酸鹽[21]等項目的檢測均取得了較好的結果。本文使用課題組自主設計并制備的快速水蒸氣蒸餾儀提取食品中氰化物,再用流動注射儀檢測,將檢測流程由原先的4 h~6 h縮短至30 min以內,節省了人力物力,提高了檢測效率,方法檢出限、準確度和精密度均滿足實際檢測要求。
1 試驗部分
1.1 儀器試劑
1.1.1 儀器
Seal Auto Analyzer 3型連續流動注射儀、XY-2型自動進樣器:德國Seal公司;Milli-Q型超純水系統:美國密理博公司;ME203型分析天平(感量0.001 g):瑞士梅特勒-托利多公司;快速水蒸氣蒸餾儀:天津海關動植物與食品檢測中心、天津大學精密儀器與光電子工程學院合作研發;UV-2450型分光光度計:日本島津公司;450-GC型氣相色譜儀:美國布魯克公司。
1.1.2 試劑
氫氧化鈉、鹽酸、乙酸、異煙酸、檸檬酸、硫酸鋅、酚酞、二甲基巴比妥酸、鄰苯二甲酸氫鉀、酒石酸、乙酸鋅、吡唑酮、磷酸氫二鈉、磷酸二氫鉀、磷酸、氯胺T:均為分析純,國藥集團化學試劑有限公司。
水中氰成分分析標準物質 GBW(E)080115:50.0 μg/mL,中國計量科學研究院。
氰化物標準中間液:1.0 μg/mL,吸取1 mL標準物質,用2 g/L氫氧化鈉溶液定容至50 mL。
流動注射儀所需試劑按照Seal公司自帶方法配制;分光光度法、氣相色譜法所需試劑按照GB5009.36-2016要求配制。
1.2 流動注射儀工作條件
進樣速率30個/h;進樣清洗比(時間)3∶1;蒸餾溫度135℃。
1.3 試驗步驟
1.3.1 樣品前處理
將樣品粉碎或勻漿,迅速稱取20g(精確到0.001g)待測樣品置于500 mL反應瓶中,加入200 mL去離子水,迅速安裝至快速水蒸氣蒸餾儀,并將冷凝管下端插入盛有10 mL濃度為20 g/mL氫氧化鈉溶液的三角瓶中,室溫(25℃)放置10 min。向反應瓶中加入20 mL濃度為100 g/L的乙酸鋅溶液和2 g酒石酸后,迅速將反應瓶安裝于快速水蒸氣蒸餾儀,設定接收液質量達到240 g為蒸餾終點,并啟動加熱程序。蒸餾結束后,將接收液轉移至250 mL容量瓶,定容。
1.3.2 標準工作曲線配制
分別吸取 0、0.10、0.25、0.50、1.00、2.50、5.00 mL 氰化物標準中間液于10 mL具塞比色管,用2 g/mL氫氧化鈉溶液定容,所配置的氰化物標準工作溶液的質量濃度分別為0、0.010、0.025、0.050、0.100、0.250、0.500 μg/mL。分別取標準系列溶液5 mL,用流動注射儀進行測定,以標準系列溶液為橫坐標,以峰高為縱坐標,繪制標準曲線。
2 結果與討論
2.1 試驗條件的選擇及優化
2.1.1 快速水蒸氣蒸餾前樣品浸泡時間的選擇
食品樣品中氰甙并不穩定,粉碎或勻漿后需要及時進行前處理。采用快速水蒸氣蒸餾前,樣品需室溫(25℃)中浸泡一段時間使氰甙水解。試驗過程中,選取鮮木薯、苦杏仁原油、亞麻籽粉和烏豆作為目標樣品,考察了浸泡時間對檢測結果的影響,如表1所示。

表1 不同浸泡時間下提取氰化物檢測結果差別Table 1 Difference of cyanide extraction test results under different soaking time mg/kg
對于3種不同類型食品,浸泡時間分別為10 min、30 min、1 h、2 h,所得檢測結果均在10%誤差范圍內,并無顯著性差別,為提高試驗效率,故選擇浸泡時間為10 min。
2.1.2 水蒸氣蒸餾速度的選擇
快速水蒸氣蒸餾儀可根據實際需求選擇蒸氣量,蒸氣量的大小直接決定蒸餾時間的長短。試驗過程中,選取鮮木薯、苦杏仁原油、亞麻籽粉和烏豆作為目標樣品,在浸泡時間均為10 min的條件下,分別選擇高、中、低3種不同大小蒸氣量對待測樣品進行氰化物提取,所得結果無顯著性差異,故選擇較高蒸氣量開展后續試驗,樣品提取時間約8 min~10 min,具體結果見表2。
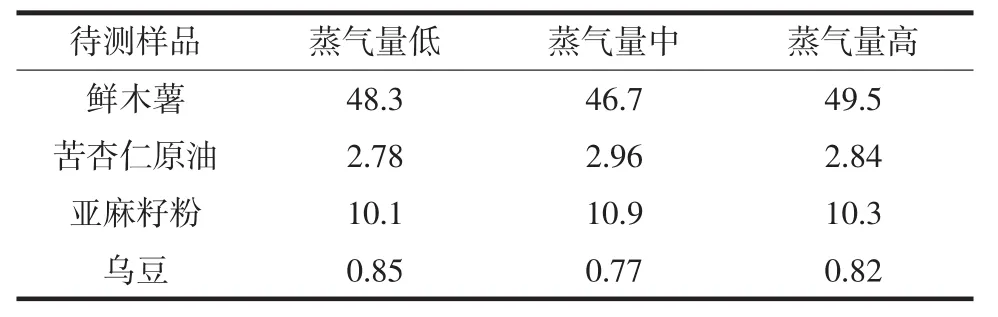
表2 不同蒸氣量下提取氰化物檢測結果差別Table 2 Differences of cyanide extraction test results under different vapor volumes mg/kg
2.2 線性方程及方法檢出限
用連續流動注射儀對1.3.2中所述氰化物標準工作溶液進行測定,以標準工作液濃度為橫坐標(X,μg/mL),以峰高為縱坐標(Y),進行線性擬合,得線性方程Y=498 321X+6 852.5,線性相關系數為0.999 6。對空白溶液進行6次重復測定,以測定值3倍標準偏差計算方法檢出限,得方法檢出限為0.001 1 mg/L。當稱樣量為20 g,定容體積為250 mL時,待測樣品中氰化物的檢測限為0.014 mg/kg(以氰離子計)。
2.3 精密度測試
以鮮木薯、苦杏仁原油、亞麻籽粉和烏豆作為目標樣品,每個樣品按照前述方法全流程重復測定6次,計算結果的精密度,結果見表3。
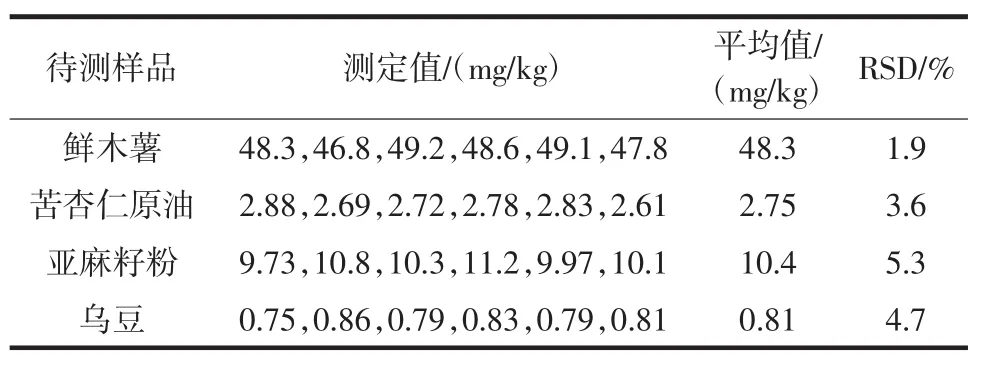
表3 精密度試驗結果Table 3 Precision test results
由表3可見,4個樣品檢測結果相對標準偏差(relative standard deviation,RSD)為 1.9%~5.3%,表明本方法精密度較好。
2.4 加標回收測試
以鮮木薯、亞麻籽粉和烏豆作為目標樣品,分別加入高、中、低3種不同濃度的氰化物標準中間液,每個樣品按照前述方法全流程重復測定6次,計算每個樣品的平均加標回收率,結果見表4。

表4 加標回收試驗結果Table 4 Recovery test results
由表4數據可以看出,3個不同濃度的樣品平均加標回收率為96.5%~104%,表明本方法具有較高的準確度。
2.5 本方法與國標方法結果比對
用國家標準GB 5009.36-2016檢測方法分別對鮮木薯、苦杏仁原油、亞麻籽粉和烏豆進行檢測,所得結果與本文方法進行比對,結果見表5。
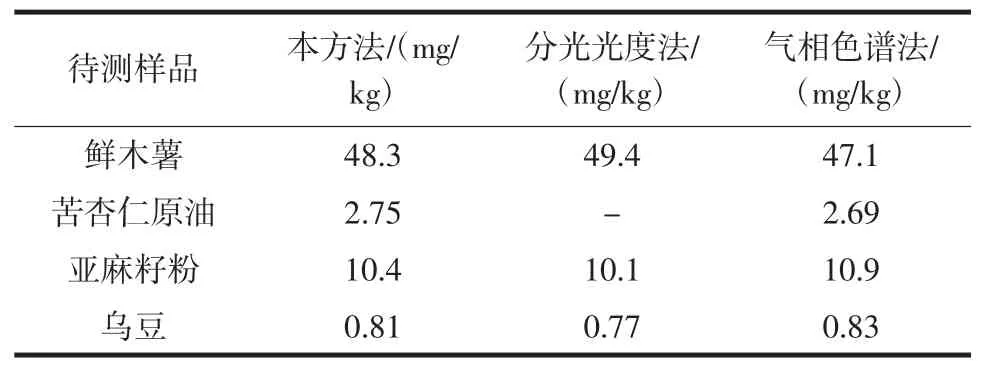
表5 方法比較Table 5 Method comparison
在進行苦杏仁原油檢測時,發現國標第一法顯色步驟產生白色渾濁,無法進行正常顯色反應。查閱文獻得知[22-23],苦杏仁原油中氰化物來源于苦杏仁苷,而苦杏仁苷分解后不僅會產生氫氰酸,同時也會產生苯甲醛,苯甲醛同樣會通過水蒸氣蒸餾的過程進入待測液,在顯色反應時與吡唑啉酮產生白色渾濁,從而影響正常顯色反應的發生。本文所述流動注射分析采用異煙酸-巴比妥酸作為顯色劑,巴比妥酸不會與苯甲醛發生類似反應,試驗中可正常發生顯色反應,因而檢測結果不受苯甲醛影響,與氣相色譜法檢測結果相一致。從表5可以看出,本方法檢測結果與分光光度法、氣相色譜法結果具有較好的一致性。
2.6 硫氰酸鹽干擾測試
試驗中以亞麻籽粉和烏豆為目標樣品,比較了本方法和氣相色譜法抗硫氰酸鹽干擾的情況,結果見表6。
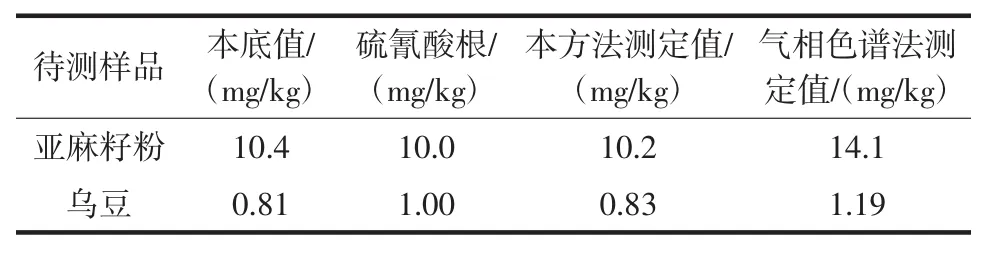
表6 硫氰酸鹽干擾試驗Table 6 Thiocyanate interference test
通過試驗結果可以看出,硫氰酸鹽存在下本方法檢測結果無明顯變化,說明方法有較好的抗硫氰酸鹽干擾的能力;而當硫氰酸鹽存在時,氣相色譜法檢測結果明顯偏高,檢測結果影響較大。分析原因如下:本方法采用水蒸氣蒸餾作為前處理,可將揮發性較強的氫氰酸蒸出,而硫氰酸揮發性較弱,留在蒸餾液中,從而將氰離子和硫氰酸根離子分開,去除干擾。氣相色譜法是在頂空瓶中將氰離子在酸性條件下用氯胺T衍生為氯化氰,在相同的條件下,硫氰酸根可發生相同的衍生化反應,從而使檢測結果普遍偏高。
3 結論
本文建立了快速水蒸氣蒸餾-流動注射分析儀聯用測定食品中氰化物含量的檢測方法。快速水蒸氣蒸餾儀的使用,能夠有效提高工作效率;流動注射分析儀的使用,簡化了試驗操作,節約了人力,提高了試驗的穩定性。數據結果表明:本方法檢出限、定量限、精密度和準確度均能夠滿足日常食品檢測需求;采用本方法和國標分光光度法、氣相色譜法同時測定4種不同食品樣品中氰化物的含量,所得結果數據無顯著性差異。與國標方法相比,本方法可有效避免樣品基質中苯甲醛、硫氰酸鹽等對試驗結果的干擾,方法適用性較廣。本方法全流程檢測時間僅需約30 min,檢測結果準確度、穩定性良好,在分析大批量食品樣品時具有較大優勢。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
