熱堿致魔芋膠與黃原膠共混凝膠的顯微結構與流變規律
2020-10-15 08:51:10李培源李曉飛李安琪余文艷郭綽楊曦郭玉蓉
中國農業科學 2020年18期
關鍵詞:體系
李培源,李曉飛,李安琪,余文艷,郭綽,楊曦,郭玉蓉
(陜西師范大學食品工程與營養科學學院,西安 710119)
0 引言
【研究意義】魔芋為天南星科魔芋屬植物,其球狀塊莖中主要成分為魔芋葡甘露聚糖,簡稱魔芋膠[1]。魔芋膠是一種性質優良的水溶性膳食纖維,不被人體的消化酶分解,但在小腸下部和大腸中被腸道菌群分解為葡萄糖和甘露糖,可調節人體膳食營養平衡[2-3]。魔芋膠凝膠食品長期以來一直被認為是健康和低熱量的食品[4]。通常,魔芋膠在熱堿處理下能形成熱不可逆凝膠,但熱堿所致的魔芋膠凝膠存在易析水、凝膠強度不足等問題,限制了魔芋膠凝膠在食品領域的進一步應用[5-8]。黃原膠是以玉米淀粉、蔗糖等為主要原料在甘藍黑腐病野油菜黃單胞菌作用下,經好氧發酵產生的一種高黏度水溶性微生物胞外多糖[9-10]。黃原膠完全水化后可得到高黏度的溶液[11],但黃原膠在單獨存在的情況下不能形成凝膠。有證據表明[12],魔芋膠與黃原膠在一定條件下復配能形成強度較高的凝膠,顯現出良好的協同增效性。然而,這種凝膠是熱可逆型凝膠,加熱時,凝膠轉變為溶膠,限制了這種凝膠在蒸煮、焙烤食品中的應用[13-16]。因此,探索魔芋膠和黃原膠復合體系在堿性條件下共熱的凝膠化作用,以期將魔芋膠凝膠網絡和魔芋膠與黃原膠協同凝膠網絡合二為一,構建出兼具兩種凝膠優點的復合凝膠,可為魔芋膠凝膠相關食品的設計提供理論依據和技術參考。【前人研究進展】ZHOU等[17]研究發現魔芋膠溶液與Na2CO3混合后并加熱到70℃以上時,魔芋膠分子立即發生脫乙酰反應,誘導溶液發生凝膠化,進一步研究發現 KGM 鏈的分子間聚集不僅與氫鍵有關,還與疏水相互作用緊密相關。因此,降低魔芋膠凝膠溫度時,疏水作用力減弱,魔芋膠凝膠強度也降低[18]。此外,由于乙酰基是賦予魔芋膠良好水溶性的關鍵因素,因此脫去乙酰基后,魔芋膠分子及其凝膠網絡的持水能力大大減弱,引起凝膠析水[19]。迄今為止,已有少量研究致力于改善魔芋膠凝膠的這些缺點,包括增加魔芋膠濃度、優化凝膠形成條件等。然而,這些方法雖然可以起到一定的效果,但并未從根本上解決這一問題。近年來,有關混合多糖體系的研究已經得到迅速發展。與單一多糖體系相比,混合多糖體系的流變特性更具有可調控性[20]。一般地,可以通過調控混合多糖凝膠網絡的形成和構建方式達到調整宏觀凝膠體系物理特性的目的。例如,LI等[21]研究了葡萄糖酸內酯誘導的海藻酸鈉和結冷膠混合體系的凝膠化作用,發現通過調整兩種多糖各自凝膠網絡的交聯密度,可以實現整個凝膠體系物理特性的調控,包括凝膠硬度、溶脹性、熱穩定性等。此外,李曉飛等[22]首次研究了堿性加熱條件下,魔芋膠與黃原膠復合凝膠的形成過程,結果發現固定魔芋膠濃度為2.0%的前提下,在0—1.5%范圍內隨黃原膠添加量增加,復合凝膠破裂強度呈增加趨勢,原因歸結于熱堿誘導的魔芋膠網絡和黃原膠發生了協同結合作用。然而,有關熱堿條件下魔芋膠和黃原膠協同結合的機理尚缺乏深入研究。【本研究切入點】本研究以魔芋膠和黃原膠復合體系為基質,研究熱堿處理下魔芋膠凝膠網絡的形成過程以及隨后冷卻過程中魔芋膠網絡與黃原膠的協同結合作用,探討熱堿處理對魔芋膠和黃原膠復合體系最佳協同比例的影響。【擬解決的關鍵問題】揭示魔芋膠與黃原膠復合體系在熱堿處理以及隨后冷卻過程中兩種凝膠網絡的形成過程及相互作用。同時,構建兼具兩種凝膠優點的復合凝膠,為魔芋膠凝膠的物理改性提供理論依據和技術參考。
1 材料與方法
試驗于 2019年在陜西師范大學食品科學與營養工程學院進行。
1.1 材料與試劑
魔芋膠和黃原膠均購自上海源葉生物科技有限公司,純度為90%以上;無水碳酸鈉、檸檬酸,購自天津市科密歐化學試劑有限公司,均為分析純。
1.2 儀器與設備
電子天平 PL-203,梅特勒-托利多儀器(上海)有限公司;EYELA OSB-2100恒溫水浴鍋;電熱鼓風干燥箱101-0A,天津心雨儀器儀表有限公司;TA. XT.Plus質構儀,英國stable micro system公司;環境掃描電子顯微鏡 Quanta 200,FEI公司;冷凍干燥機FD-1A-50,北京博醫康實驗儀器有限公司;AR-G2流變儀,美國TA公司。
1.3 方法
1.3.1 樣品制備 準確稱取25.0 g魔芋膠粉末溶解于1.0 L的去離子水中,一邊添加魔芋膠粉末一邊快速攪拌,隨后將溶液置于90℃水浴鍋中恒溫加熱4 h,并不斷攪拌至魔芋膠完全溶解,得到 2.5%魔芋膠溶液。黃原膠母液制備方法同上。稱取25.0 g黃原膠粉末溶解在1.0 L的去離子水中,一邊添加黃原膠粉末一邊快速攪拌,隨后將溶液置于90℃水浴條件下不斷攪拌至完全溶解[23]。
1.3.2 凝膠制備 在魔芋膠和黃原膠總濃度為2.0%的條件下,改變魔芋膠與黃原膠的比例,制備不同魔芋膠和黃原膠配比的復合體系。參照表1,兩者總體積固定為160 mL,按照10∶0到0∶10的體積比改變魔芋膠母液和黃原膠母液的混合比例,之后各體系分別加入30 mL去離子水,并置于90℃水浴中加熱攪拌至魔芋膠和黃原膠混合均勻。將4.0 g無水碳酸鈉用10 mL去離子水溶解,緩慢加入到復合體系中,并快速攪拌,此時各個復合體系的總體積為200 mL。待復合體系混合均勻后,轉入200 mL燒杯,封上PE保鮮膜,將燒杯在90℃水浴中加熱2 h,取出燒杯,室溫下自然冷卻。室溫平衡12 h后,測定各個凝膠的破裂強度,找到最佳配比[24]。采用該方法制備得到的復合凝膠命名為熱堿協同處理的凝膠。
此外,本試驗進一步制備了不同比例的魔芋膠和黃原膠復合體系,但該體系不在 90℃水浴條件下加熱,即不誘導魔芋膠網絡形成。參照表1,在總體積為160 mL的前提下,改變魔芋膠母液和黃原膠母液的體積比,制備不同混合比例的復合體系。將該復合體系轉入200 mL燒杯中,置于90℃水浴中加熱,并持續攪拌,使魔芋膠和黃原膠充分混合。各體系中分別加入30 mL溫度為90℃的去離子水,繼續攪拌均勻。之后,稱取4.0 g碳酸鈉粉末溶于10 mL去離子水中,充分溶解后,緩慢加入復合體系中,并劇烈攪拌,使體系混合均勻。立刻取出燒杯,自然冷卻至室溫。室溫平衡12 h后,測定凝膠強度。這一方法制備得到的復合凝膠命名為堿處理凝膠。
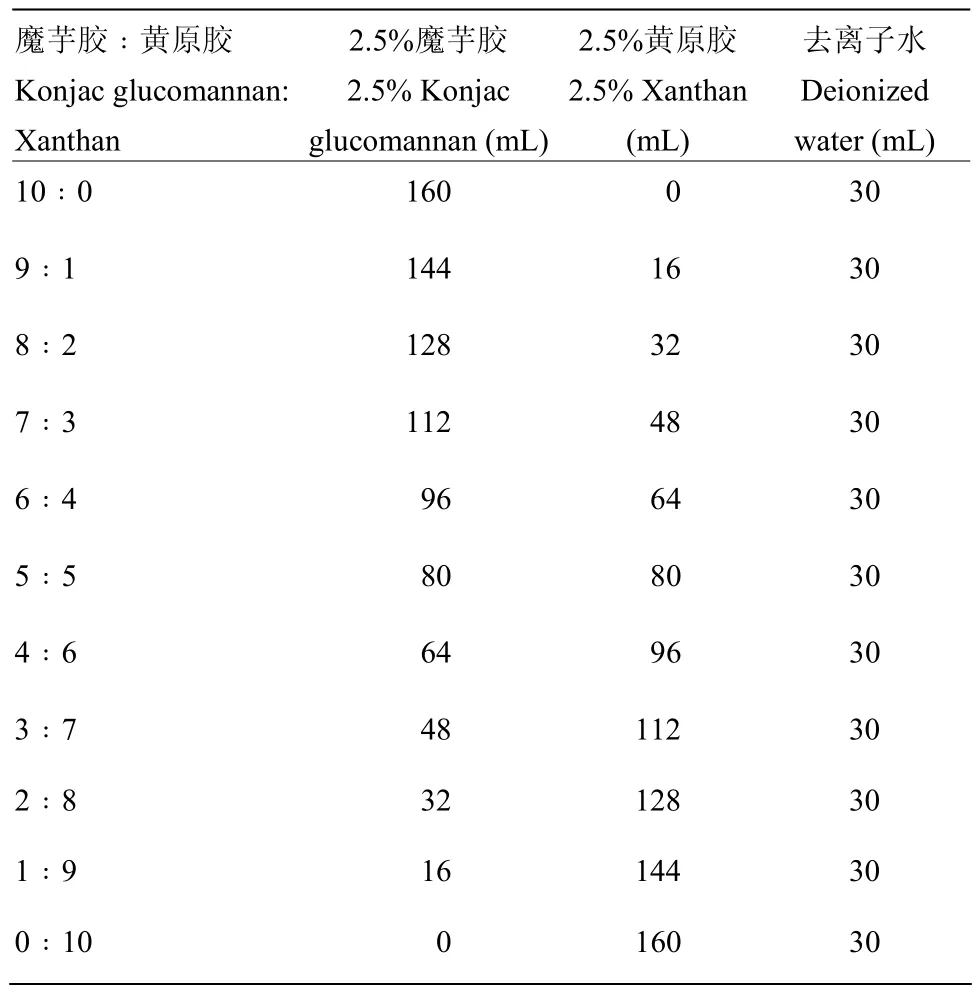
表1 樣品制備各組分添加量Table 1 Addition amount of each component for sample preparation
1.3.3 凝膠破裂強度測定 凝膠平衡12 h后,采用質構儀測定凝膠破裂強度。參數設定為:TPA壓縮模式,探頭P 0.5(直徑0.5 cm的圓柱狀平頭探頭),測試前速度:1.0 mm·s-1;測試速度:1.0 mm·s-1;測試后速度:1.0 mm·s-1;感應力:2.0 g;凝膠破裂點所對應的力記為凝膠強度[25],每個測定重復3次。
1.3.4 凝膠微觀形貌觀察 參照上述步驟制備凝膠并于室溫下平衡12 h后,將所有凝膠切割成邊長約為2 cm的立方塊,并迅速將凝膠浸沒于液氮中速凍。液氮浸沒約3 min后,將凍住的凝膠立刻轉移至真空冷凍干燥機中,在-50℃條件下凍干48 h。將凍干的凝膠折斷,斷面噴金,采用Quanta 200 環境掃描電鏡觀察凝膠斷面形貌,觀察時選擇高真空模式,高壓為20.0 kV[26]。
1.3.5 溶脹特性 參照 1.3.2中的步驟制備復合凝膠。分別采用去離子水和2.0%的檸檬酸溶液浸泡(pH 2.06)復合凝膠。浸泡 12 h后取出凝膠,參照 1.3.3中的方法測定凝膠破裂強度。此外,參照1.3.4中的步驟,觀測微觀形貌[27]。
1.3.6 凍融穩定性 參照 1.3.2中的步驟制備復合凝膠,將所有復合凝膠置于-18℃冰箱中冷凍12 h,取出凝膠,室溫下自然解凍6 h,參照1.3.3中的方法測定凝膠強度。此外,也測定凍融后凝膠的析水率。凝膠冷凍前的質量記為m1,凝膠凍融后將析出的水分除去,剩下的質量記錄為m2。析水率可由下式計算:
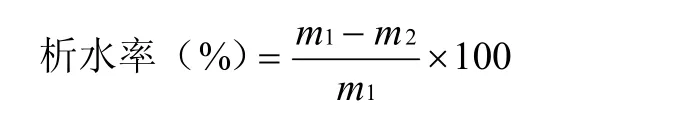
1.3.7 流變學性質 時間掃描:魔芋膠和黃原膠復合體系及碳酸鈉溶液均按照1.3.2的方法制備并混合。按照表1添加配置好的2.5%魔芋膠母液、2.5%黃原膠母液和去離子水,并和 Na2CO3溶液混合,加入燒杯中快速攪拌均勻,制得魔芋膠與黃原膠比例分別為10∶0、9∶1、7∶3、5∶5的體系。隨后,采用AR-G2型流變儀測定各復合體系的流變特性,所選夾具為直徑為20 mm的糙面平行板。操作步驟如下:立刻移取2 mL混合溶液置于流變儀樣品臺上,平行板間距設置為 1 mm,并在平行板四周涂抹低密度硅油,防止測試過程中由于樣品水分蒸發而造成較大誤差。之后,在90℃條件下進行120 min的時間掃描,監測儲能模量G'和損耗模量G''的變化趨勢,應變范圍為1%(線性粘彈區內),頻率為1 Hz。
降溫掃描:時間掃描程序結束后,立刻在90—20℃范圍內進行降溫掃描,降溫速率為2℃·min-1,應變為1%,頻率為1 Hz[28]。
頻率掃描:當樣品溫度降低至20℃后,在該溫度條件下平衡10 min,之后在1—100 rad·s-1頻率范圍內掃描,應變為1%。
方程擬合:為了進一步分析凝膠在相同堿濃度(2%)及溫度(90℃)下的凝膠化過程,將上述時間掃描的G'匯總,并采用如下方程進行擬合[29]:

式中,G′sat為無限的加熱時間下,G′的極限值;k為凝膠化速率參數;x為時間;y為儲能模量G′。
1.3.8 數據處理 上述所有測定均重復3次,每次測試均需更換樣品。所有圖表均使用Origin 2018軟件進行繪制,利用IBM SPSS22.0軟件進行差異顯著性分析,P<0.05表示差異顯著。
2 結果
2.1 凝膠強度及外觀
破裂強度是反映凝膠網絡交聯密集程度的重要指標。圖 1-A為 2.0%的魔芋膠溶膠在熱堿條件下(90℃)加熱0—3 h的強度測定結果。如圖所示,隨加熱時間增加,凝膠強度也逐漸增強,加熱2 h后,凝膠強度雖然略有增加,但變化不顯著(P>0.05)。由圖1-B可知,加熱時間為0 h時,魔芋膠不能形成凝膠,加熱0.5 h后可形成明顯的凝膠。基于這一結果,后續試驗選擇2 h作為凝膠熱處理的時間。

圖1 不同魔芋膠和黃原膠比例復合凝膠強度及凝膠外觀Fig. 1 Gel strength and gel appearance of different konjac glucomannan to xanthan ratios
將不同比例的魔芋膠和黃原膠混合,在熱堿協同處理2 h和堿處理后(未在90℃條件下加熱)分別測定凝膠強度,結果見圖 1-C。如圖所示,對于堿處理的凝膠,魔芋膠與黃原膠比例為5∶5時,凝膠破裂強度最高,約為1 400 g。對于熱堿協同處理的凝膠,魔芋膠與黃原膠比例為7∶3時凝膠強度最大,約為1 200 g。可能是由于熱堿協同處理過程中,多糖分子發生部分降解,影響了復合凝膠的強度[30]。魔芋膠與黃原膠比例為10∶0和9∶1時(圖1-D、E),堿處理條件下凝膠無法形成,隨黃原膠比例增加,凝膠呈現先增強后降低的趨勢,和圖1-C結果一致。熱堿協同處理后,所有凝膠均呈棕色,且凝膠色澤隨黃原膠比例增加逐漸變淺,當魔芋膠與黃原膠比例為1∶9和0∶10時無法形成凝膠,原因可能是黃原膠的存在降低了魔芋膠凝膠網絡形成。
2.2 凝膠形貌
凝膠微觀形貌與其強度密切相關[31]。由圖2可知,不同比例的復合凝膠均形成了網絡結構,但魔芋膠凝膠(魔芋膠∶黃原膠=10∶0)的微觀形貌為層狀結構,網絡不明顯;隨黃原膠加入,凝膠開始出現網狀結構,當魔芋膠與黃原膠比例為7∶3時,網絡最為致密。當黃原膠比例繼續增加時,網絡結構變得疏松多孔。該結果與圖1測得的結果相符,表明凝膠網絡結構越致密,凝膠強度越大。
2.3 去離子水與檸檬酸溶液浸泡對凝膠結構的影響
由于凝膠制備過程中加入了2.0%的碳酸鈉,可能造成凝膠存在明顯的堿味,因此使用過程中需要將堿味去除。采用去離子水和檸檬酸溶液浸泡處理去除凝膠的堿味,并研究浸泡后凝膠強度及微觀形貌的變化。如圖3-A所示,去離子水浸泡后凝膠強度略有下降,但魔芋膠與黃原膠比例為7∶3時,凝膠仍具有最高的強度,表明去離子水浸泡并未改變復合凝膠的最佳協同比例。由圖3-B可知,魔芋膠與黃原膠比例為7∶3時,凝膠網絡結構最為致密。2.0%檸檬酸溶液浸泡后,魔芋膠與黃原膠比例為7∶3時凝膠強度也最大,且凝膠網絡結構也最致密(圖3-C、D)。然而,與去離子水浸泡的凝膠相比,檸檬酸浸泡后的凝膠網絡更疏松,結構更松散,可能是因為檸檬酸與凝膠中的碳酸鈉發生反應生成了二氧化碳,擾亂了凝膠原有的網絡結構。
2.4 凝膠的凍融穩定性
將復合凝膠置于-18℃中冷凍12 h,取出后于室溫下解凍,測定凝膠破裂強度、析水率及凝膠微觀形貌。由圖4-A、B可知,當魔芋膠與黃原膠比例為10∶0、9∶1和8∶2時,凍融后凝膠強度比冷凍之前有所上升,原因可能是冷凍過程中水分子結晶,迫使魔芋膠分子聚集形成穩定的交聯,室溫條件下解凍時,交聯也不會解聚,引起凝膠強度增加。然而,魔芋膠與黃原膠比例為7∶3、6∶4、5∶5時,凍融后凝膠強度比冷凍前有所下降,原因可能是黃原膠比例增加,凝膠已經形成了較強的網絡結構,冷凍過程中由于水分結晶,破壞了凝膠的原有網絡,造成凝膠強度降低。此外,隨黃原膠比例增加,凝膠析水作用逐漸降低(圖4-C)。魔芋膠與黃原膠比例為10∶0和9∶1時,凝膠形貌為板狀,網絡結構不明顯,魔芋膠與黃原膠比例為8∶2、7∶3、6∶4、5∶5時網絡結構明顯,表明魔芋膠比例越高,復合凝膠的凍融穩定性越差(圖4-D)。

圖2 不同魔芋膠和黃原膠比例對復合凝膠微觀形貌的影響Fig. 2 SEM images of the composite gels with different konjac glucomannan to xanthan ratios
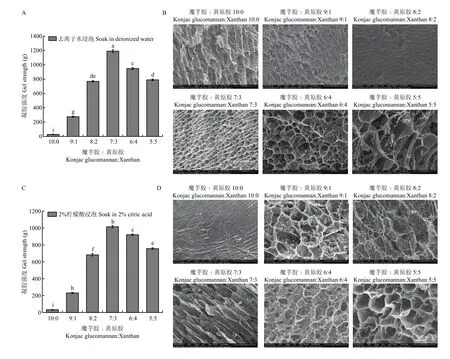
圖3 水溶液浸泡對不同比例的魔芋膠和黃原膠復合凝膠破裂強度及微觀形貌的影響Fig. 3 Effects of water immersion on the rupture strength and SEM images of the composite gels with different konjac glucomannan to xanthan ratios
2.5 流變特性
2.5.1 時間掃描 混合溶膠在90℃持續加熱時,可觀察到隨時間增加,G'和 G''均呈顯著增加趨勢,0—1 200 s內G'增加速率顯著高于G'',表明凝膠網絡迅速生成。3 600 s之后,雖然G'和G''仍在繼續增加,但增加趨勢較為緩慢,說明凝膠結構發展比較完善。對于2.0%的魔芋膠體系,在240 s左右時G'和G''出現了交點,但在其他3個比例條件下,整個掃描過程中均未能觀察到G'和G''的交點,可能是因為黃原膠比例增加后,由于黃原膠自身的分子間纏結作用,使得整個體系表現出 G'大于 G''的特征(圖5)。
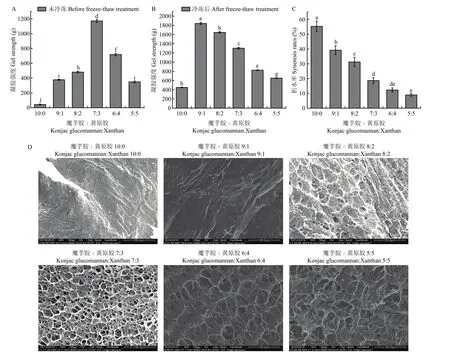
圖4 凍融處理對不同比例的魔芋膠和黃原膠復合凝膠破裂強度、析水率及微觀形貌的影響Fig. 4 Effects of freeze-thaw treatment on the rupture strength and syneresis rates and micro-morphologies of the composite gels with different konjac glucomannan to xanthan ratios
2.5.2 方程擬合 為了進一步分析凝膠在相同的堿濃度(2.0%)及溫度(90℃)條件下的凝膠化過程,將上述時間掃描的 G' 匯總,并采用如下方程進行擬合[29]:,結果見圖6。擬合的G'sat和k值見表2,所有擬合曲線的R2均在0.98以上,表現出較高的擬合精度。
由表2和圖6可知,2.0%魔芋膠的G'sat和k值最大,隨黃原膠添加量增加,G'sat和k都降低,說明黃原膠添加不僅降低了魔芋膠網絡最終的強度,而且減緩了魔芋膠凝膠網絡的形成速率。
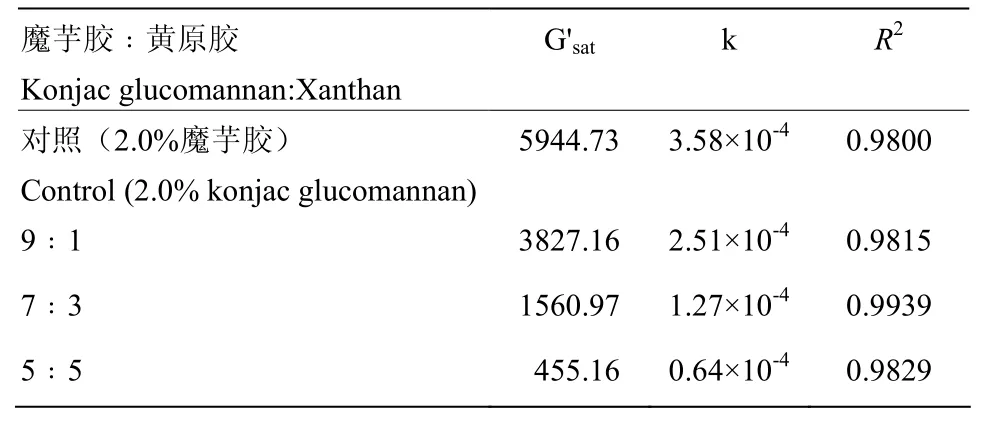
表2 不同魔芋膠/黃原膠比例凝膠的G'一級動力學擬合方程參數Table 2 Parameters of the fitted equation of first order kinetics of G' of the composite gels with different konjac glucomannan to xanthan ratios

圖5 不同魔芋膠和黃原膠比例的復合體系在90℃條件下時間掃描結果Fig. 5 Time sweep curves of the prepared konjac glucomannan/xanthan gels with fixed total gum concentration but varying konjac glucomannan to xanthan ratios at 90℃

圖 6 不同魔芋膠和黃原膠混合比例的復合體系 G'的變化曲線及方程擬合Fig. 6 G' evolution process of the composite gels with different konjac glucomannan to xanthan ratios
2.5.3 溫度掃描 魔芋膠與黃原膠協同凝膠作用與溫度有關,熱堿協同處理2 h后,2.0%的魔芋膠體系隨溫度降低,G'和G''呈不斷下降趨勢,說明魔芋膠凝膠強度也隨之降低。由圖7-B—D可知,G'在90—60℃范圍內呈現降低趨勢,60℃以后G'逐漸上升,可能是由于溫度從90℃降低至60℃時,維系魔芋膠凝膠網絡的疏水作用力變弱,從而導致凝膠網絡弱化。60℃以后G'開始增加,可能是該溫度下黃原膠和魔芋膠網絡發生了協同結合作用,從而加固了凝膠體系,溫度越低,協同結合作用越強[29]。當溫度從 90℃降低至60℃時,G'表現出緩慢增加的趨勢,然而當溫度降低至60℃以后,G'迅速上升,表明黃原膠和魔芋膠協同作用發生的起始溫度為60℃(圖7-E)。這一結果也證實了熱堿協同凝膠降溫過程中G'的增加是由于黃原膠和魔芋膠網絡發生了協同凝膠化。當溫度從90℃降低至46℃時,G'呈現緩慢增加趨勢,此后隨溫度降低,G'迅速上升(圖7-F)。圖7-E、F對比說明碳酸鈉添加改變了魔芋膠與黃原膠凝膠體系協同作用的起始溫度,在不加堿條件下協同化溫度為46℃,而加入堿以后兩者協同化溫度上升至60℃。
2.5.4 頻率掃描 頻率掃描可得到凝膠儲能模量 G'和損耗模量G''隨振蕩頻率變化的關系,是評估凝膠狀態的重要指標[32]。由圖8可知,各比例條件下,凝膠體系在 1—10 rad·s-1的頻率范圍內模量幾乎不隨頻率升高而增加,說明已經形成較強的凝膠。其中,魔芋膠與黃原膠比例為7∶3時,G'值最大,而魔芋膠與黃原膠比例為10∶0的凝膠G'最低,這一結果和凝膠強度測定結果一致。
3 討論
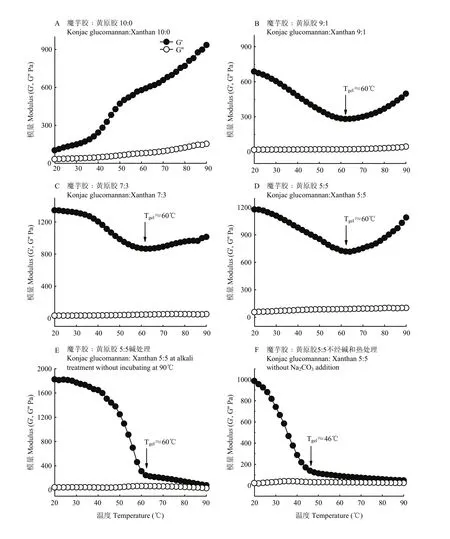
圖7 不同魔芋膠和黃原膠比例下復合凝膠體系的溫度掃描結果Fig. 7 Gel evolution process (G') of the composite gels with different konjac glucomannan/xanthan ratios during cooling

圖8 不同魔芋膠和黃原膠比例下復合凝膠體系的頻率掃描結果Fig. 8 Frequency sweep curves of the composite gels with different konjac glucomannan to xanthan ratios
研究表明[18],魔芋膠在堿性條件下形成凝膠的速率與魔芋膠濃度、堿濃度、加熱溫度、魔芋膠濃度與堿濃度之比有關。魔芋膠濃度越高,堿添加量越多、加熱溫度越高,魔芋膠去乙酰化速率越快,凝膠化速率也越高。本研究發現魔芋膠與黃原膠混合體系在最初的2 400 s內彈性模量和黏性模量均呈現迅速增加的趨勢,2 400 s后,兩者的增加速率逐漸變緩。然而,整個時間掃描過程中,基本未監測到G'和G''的交點,可能是由于混合體系加熱溫度較高,且碳酸鈉添加量也較高,導致混合體系凝膠速率過快,在時間掃描程序開始運行之前,魔芋膠已經發生脫乙酰基反應,形成微弱的網絡結構。因為G'是反應凝膠體系彈性特征的重要參數,因此本研究進一步通過方程擬合的方法探究熱堿協同處理對魔芋膠與黃原膠混合體系的影響。結果發現,隨魔芋膠與黃原膠比例降低,混合體系的凝膠速率常數k以及G'sat也降低,可能是體系中魔芋膠濃度降低所致。
加熱2 h后,所有的混合體系均能形成凝膠。然而,當冷卻混合凝膠體系至室溫時,凝膠強度極大增加,顯然是由于混合凝膠體系在冷卻過程中,黃原膠分子與魔芋膠網絡發生了協同作用[22]。因此,為進一步揭示這種協同作用的機制,本研究對混合凝膠體系進行降溫掃描。結果發現,當溫度從90℃降低至60℃時,所有的混合凝膠體系的G'均表現出降低的趨勢,這可能是因為隨溫度降低,魔芋膠分子間的疏水作用力減弱,導致魔芋膠三維網絡松散。然而,當溫度進一步降低時,混合凝膠體系的G'表現出增加的趨勢,表明黃原膠分子開始和魔芋膠網絡協同結合,加固了凝膠網絡。
黃原膠和魔芋膠最佳協同比例為4∶6(黃原膠∶魔芋膠),在此比例下,凝膠強度最大[24]。然而,黃原膠和魔芋膠最佳協同比例隨體系中離子濃度的變化而輕微變化。例如,有報道顯示[33],存在鈉離子時,黃原膠與魔芋膠最佳協同比例由4∶6偏移至5∶5,這是由于鈉離子屏蔽了黃原膠分子上微弱的負電荷,促使黃原膠分子發生聚集,減少了與魔芋膠分子發生協同結合作用的有效黃原膠含量,因此在達到最佳協同比例時需要更多的黃原膠。本研究發現,添加2.0%碳酸鈉時,黃原膠與魔芋膠最佳協同比例為5∶5。然而,在90℃條件下加熱魔芋膠和黃原膠混合體系2 h并冷卻至室溫后,最佳協同比例為7∶3(魔芋膠∶黃原膠)。原因可能是魔芋膠和黃原膠的協同結合位點數目有所變化。李曉飛等[22]研究發現,堿性條件下加熱魔芋膠和黃原膠的混合體系并冷卻時,魔芋膠分子首先發生脫乙酰基反應,形成魔芋膠凝膠網絡,隨后冷卻過程中魔芋膠網絡和黃原膠發生協同結合,進一步加固凝膠。因此,堿性條件加熱時,魔芋膠網絡首先形成,在隨后的冷卻過程中,與黃原膠協同結合位點明顯減少,在達到最大協同比例時,需要更多的魔芋膠分子參與,意味著魔芋膠與黃原膠比例也相應增加。
4 結論
在90℃條件下加熱魔芋膠和黃原膠共混體系時,形成熱不可逆凝膠網絡,當降低該體系的溫度至60℃時,黃原膠分子與魔芋膠網絡發生協同結合,增加了凝膠的彈性模量,隨溫度進一步降低,凝膠彈性模量持續增加。冷卻至室溫時,復合凝膠的最佳協同比例為魔芋膠∶黃原膠為 7∶3。經去離子水和 2.0%檸檬酸溶液浸泡后,凝膠強度均有所降低,但并未改變凝膠的最佳協同比例。凍融處理后,凝膠出現明顯的析水現象,隨黃原膠比例增加,凝膠析水作用降低。綜上,本研究發現熱、堿協同處理魔芋膠與黃原膠的混合體系可以改善單純堿法誘導的魔芋膠凝膠凍融穩定性差、凝膠強度不足等缺點。
致謝:感謝陜西師范大學化學與化工學院大型儀器共享平臺的儀器支持及管理老師在測樣過程中的指導和幫助。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
