超速離心-沉降速率法測定蛋白質分子量的實驗條件
2020-09-29 05:48:48阮梅林
實驗技術與管理 2020年6期
關鍵詞:實驗
雷 敏,劉 斌,阮梅林,羅 云
(華中科技大學 生命科學研究共享平臺,湖北 武漢 430074)
蛋白質分子量是蛋白的重要特征參數,同時是研究蛋白均一性、聚集狀態和蛋白間相互作用等特征參數的基礎。因此,準確有效地測定蛋白質分子量非常關鍵[1-2]。傳統測定蛋白質分子量的方法主要包括凝膠電泳法[3]、高效凝膠過濾色譜-多角度激光光散射法[4]、電噴霧離子化質譜法[5]以及基質輔助激光解吸電離質譜法[6-7]。其中凝膠電泳法和凝膠色譜法操作復雜,對樣品的需求量高,受樣品的形狀和尺寸影響較大,結果準確度低[8]。電噴霧離子化質譜法和基質輔助激光解吸電離質譜法是近幾年發展迅速的生物大分子測定技術,也是目前測定蛋白質分子量的主要方法[2]。近年來,由于分析超速離心法在測定蛋白質分子量的同時,可從相同數據組中同時分析出待測蛋白的均一性、聚集狀態、化學計量比和分子構象等多項特征參數,且該方法具有樣品需求量少、無需標準品、無破壞性和普適性高等諸多優勢,受到越來越多的研究者青睞[8-19]。
已有的文獻報道主要基于利用該方法測定蛋白質分子量或其特征參數,但對于檢測過程中樣品的初始濃度、離心轉速等實驗條件的改變是否會對測定結果產生影響并未提及。而研究各項實驗條件的影響并篩選出合適的參數,對于準確測定蛋白質分子量及其他特征參數具有重要的意義。因此,本文以牛血清蛋白(albumin bovine serum,BSA)和人血清免疫球蛋白(human serum immunoglobulin G,IgG)作為研究對象,在分析超速離心-沉降速率(sedimentation velocity of analytical ultracentrifugation,AUC-SV)模式下,探究了樣品初始濃度、離心轉速和樣品上樣體積等實驗條件對測定結果的影響。在優化的實驗條件下,建立了 AUC-SV 測定蛋白質分子量的方法,同時可為AUC-SV 初學者提供技術指導。
1 實驗部分
1.1 儀器
分析型超速離心機 ProteomeLab XL-I(美國Beckman 公司),型號為An-60 Ti 的4 孔轉頭,使用2-channel 樹脂中心件和藍寶石窗口組裝樣品池;紫外超微量分光光度計NanoDrop2000(Thermo 公司);基質輔助激光解析電離串聯飛行時間質譜MALDI-TOF/TOF MS 5800(美國AB SCIEX 公司)。
1.2 試劑
BSA(≥98%)標準品購自Biofroxx 公司;IgG(≥95%)標準品購自Sigma 公司;Tris(hydroxymethyl)aminomethane(試劑純,≥99%)購自Sigma 公司;磷酸鹽溶液(phosphate buffered solution, PBS)購自北京Biosharp 公司;NaCl(分析純)購自上海滬試公司。
1.3 AUC-SV
分別在分析型超速離心機中注入400 μL pH 8.0 PBS 和380 μL 樣品溶液。樣品濃度采用紫外超微量分光光度計進行測定,記錄樣品在280 nm 處的吸光度A280。設置紫外檢測波長為280 nm,掃描頻率為6 min/次,在4 ℃及40 000 r/min 的條件下離心,待樣品全部離心至池底時停止掃描和離心。使用SEDFIT軟件[20]對數據進行擬合分析后可得蛋白質分子量。
1.4 實驗條件的優化
實驗條件的優化按照樣品初始濃度、離心轉速、離心時間、離心溫度、樣品上樣體積和緩沖液體系的順序進行,每次只改變單一實驗條件,已優化的實驗條件直接應用到后續的分析中,待優化的實驗條件則參考節1.3。
1.5 質譜法測定蛋白質分子量
使用基質輔助激光解析電離串聯飛行時間質譜,在Linear High Mass Positive 模式下,以芥子酸為基質,測定未知蛋白質分子量。
2 結果與討論
2.1 樣品初始濃度的選擇
樣品性質不同及儀器性能差異均可能影響到樣品適合的初始濃度。首先測定了BSA 和IgG 初始濃度A280分別為0.1、0.4、0.7、1.0、1.2 Abs 時的分子量,結果如表1 所示。從表1 可以看出,在上述濃度范圍內,擬合得到的BSA 和IgG 分子量均與標準分子量接近,相對誤差在±5%以內。但通過對擬合得到的峰形進行分析,發現BSA 在A280為0.1 Abs 時,峰形延展較寬,擬合得到的分子量分布較實際范圍寬。而A280為1.2 Abs 時,兩者的紫外掃描誤差達到±0.05 Abs,超過儀器允許范圍(±0.02 Abs)。因此,在進行未知蛋白質分子量檢測時,建議調節樣品初始濃度A280在0.4~1.0 Abs 之間。
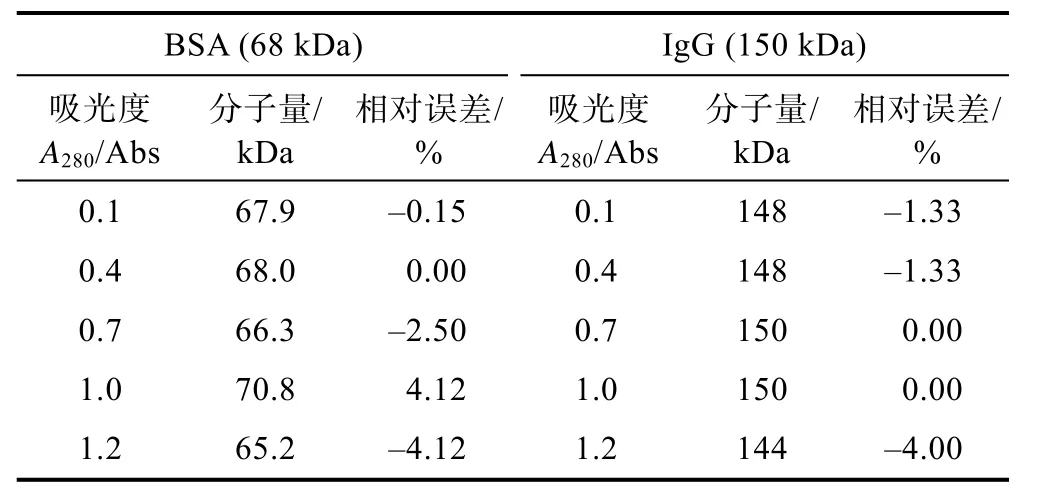
表1 不同樣品初始濃度下的分子量擬合結果
2.2 離心轉速的選擇
樹脂中心件能承受的最高轉速為40 000 r/min,因此比較了轉速在40 000、30 000、20 000 r/min 下的擬合結果,如表2 所示。在相同的離心轉速下,分子量較大的IgG 離心沉底的時間較短。在20 000 r/min時,樣品經20 h 的長時間離心后仍然未能沉底。3 個轉速條件下擬合得到的分子量與標準分子量接近,相對誤差均在±2%以內。這說明離心轉速對擬合結果影響不明顯,但轉速越低,離心時間要求越長,因此選擇40 000 r/min 可快速高效獲得蛋白質分子量。
2.3 離心時間的選擇
分析節2.2 所述(40 000 r/min)不同離心時間的數據,結果如表3 所示。BSA 離心到池底需要10 h,IgG 需要 6 h,擬合得到的分子量分別為 68.0 和150 kDa,與標準分子量一致。當離心時間較短(3~6 h)時,未離心到池底的數據經擬合得到的分子量與標準分子量也較為接近,相對誤差在±3%以內。IgG離心時間9 h 及以上,分子量相對誤差逐漸增大,由-5.33%增加到-7.33%,這是由于樣品沉底后,所采集到的數據濃度變化不明顯,有效數據減少,從而導致擬合誤差增大。因此,當樣品離心到池底時即可停止離心,同時在進行數據擬合時應注意選取擬合區間,避免導入重復的沉底數據。

表2 不同離心轉速下的分子量擬合結果

表3 不同離心時間下的分子量擬合結果
2.4 離心溫度的選擇
BSA 和IgG 在溶液狀態下穩定存在的最適溫度均為4 ℃,而SEDFIT 軟件擬合參數默認溫度為20 ℃。因此對比了在4、10、20 ℃條件下的擬合結果,如表4所示。由表4 可知,4 ℃條件下擬合得到的分子量與標準分子量一致,隨著溫度升高,相對誤差明顯增大。而根據擬合得到的峰形及峰的數量來看,BSA 和IgG在3 個溫度條件下均未出現明顯分解或聚集現象,誤差增大的原因可能是隨著溫度升高,溶液的黏度降低,樣品沉降系數變小,從而分子量測定結果偏低[21]。此外,可推斷在此實驗條件下,SEDFIT 軟件擬合時對溫度的要求并不嚴苛,因此對于未知蛋白質,可選擇在其穩定的溫度下進行離心即可。
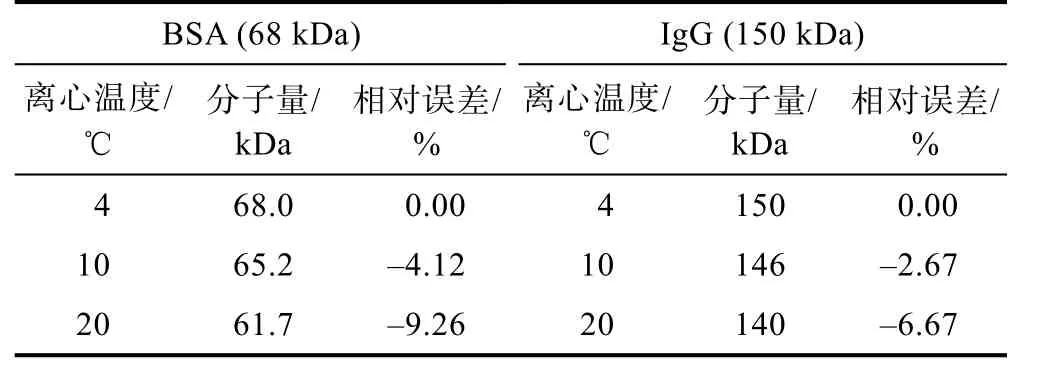
表4 不同離心溫度下的分子量擬合結果
2.5 樣品上樣體積的選擇
對上樣體積分別為100、200、380 μL 的實驗結果進行了比較,如表5 所示。隨著上樣體積的減少,BSA 和IgG 擬合得到的分子量與標準分子量之間的相對誤差都變大,且IgG 的相對誤差高于10%。這是由于樣品上樣體積減小,樣品池的有效掃描半徑和數據擬合區間縮短,導致擬合的有效數據量減少。此外,離心過程中IgG 的沉降速度更快,IgG 的有效擬合數據較BSA 少,導致擬合得到的IgG 分子量相對誤差較BSA 更大。因此建議對12mm 2-channel 樹脂中心件的上樣體積為380 μL。
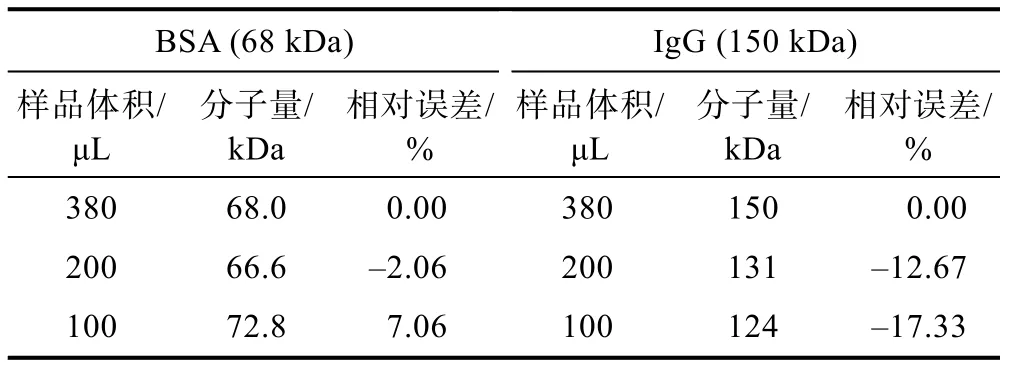
表5 不同上樣體積的分子量擬合結果
2.6 緩沖體系的選擇
比較了BSA 和IgG 在水、PBS 和150mmol/L NaCl-50mmol/L Tris 混合溶液3 種溶劑體系的實驗結果,如表6 所示。BSA 和IgG 在3 種溶液中均能穩定存在,測定結果與標準分子量接近。但已有文獻報道,測量帶電體系時需要加入小分子鹽以屏蔽溶質分子間靜電相互作用,以消除靜電相互作用對分子在溶液中運動產生的影響[22]。如對于多數等電點在4~5 之間的蛋白質,建議采用pH 范圍在7~9 之間的緩沖溶液。

表6 樣品在不同緩沖體系中的分子量擬合結果
2.7 未知蛋白質樣品分析
在上述優化的實驗條件下,以PBS 為緩沖體系,測定未知蛋白質的分子量,并與質譜測定結果進行比較,如圖1 所示,表示蛋白質濃度c 分布與沉降系數s 分布的對應關系。AUC-SV 法和MALDI-TOF/TOF質譜法測得該蛋白質分子量分別為39.9 kDa(圖1(a))和40.0 kDa(圖1(b)),兩者測定結果一致,說明該方法可用于測定其他蛋白質的分子量。
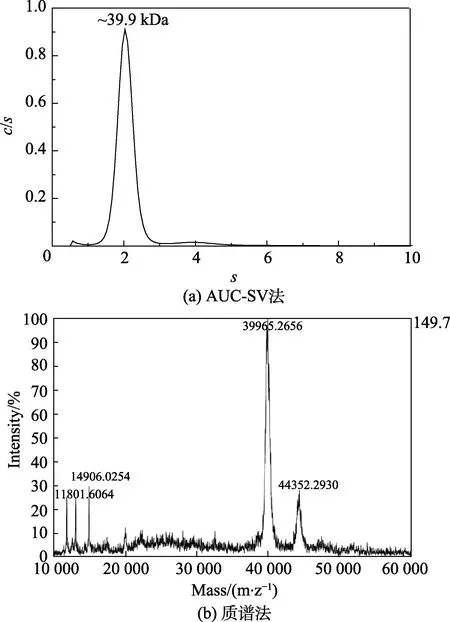
圖1 未知蛋白質分子量測定結果
3 結論
本文通過對AUC-SV 實驗條件研究發現:離心轉速在20 000~40 000 r/min 對測定結果無明顯影響,但離心轉速越大,離心時間越短,建議優先選擇40 000 r/min,以快速獲得實驗結果。樣品初始濃度、離心時間、離心溫度和緩沖液體系的適宜范圍則較寬,樣品上樣體積對測定結果的影響顯著,建議對于12mm 2-channel 樹脂中心件的上樣體積為380 μL。在優化的實驗條件下,測得未知蛋白質的分子量為39.9 kDa,與質譜法測定結果(40.0 kDa)一致。該方法具有適宜條件范圍寬、非破壞性、無需標準品和普適性高等優點,可廣泛應用于未知蛋白質分子量的測定,其主要缺點則是對樣品的純度要求較高[23],否則測定結果可能會受到雜質干擾。此外,本文詳細探討了各項實驗條件對測定結果的影響,可供AUC-SV 初學者學習借鑒。
猜你喜歡
作文·小學低年級(2025年2期)2025-02-13 00:00:00
小雪花·小學生快樂作文(2024年11期)2024-12-31 00:00:00
作文·小學低年級(2024年2期)2024-04-29 00:00:00
作文·小學低年級(2023年3期)2023-04-29 00:00:00
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
小主人報(2022年4期)2022-08-09 08:52:06
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
