基質輔助激光解吸電離-傅里葉變換離子回旋共振質譜法快速測定全血及尿液中的百草枯
2020-09-29 03:48:48羅輝泰張秋炎楊運云向章敏劉舒芹
分析測試學報 2020年9期
陳 超,羅輝泰,張秋炎,楊運云,向章敏,劉舒芹
(廣東省科學院 廣東省測試分析研究所(中國廣州分析測試中心) 廣東省原位電離質譜分析工程技術研究中心,廣東 廣州 510070)
百草枯(1,1′-二甲基-4,4′-聯吡啶二氯鹽,Paraquat)是一種聯吡啶類陽離子季銨鹽,具有高水溶性和低揮發性,是在全球范圍內被廣泛使用的農業非選擇性高效除草劑之一[1]。百草枯對人和動物具有極高的毒性,易通過皮膚、呼吸道和消化道被吸收,會對肝、肺、腎、心臟和神經系統造成不可逆的嚴重損害[2],且目前尚無特效解毒劑。自2016年起,我國已禁止生產、銷售和使用高劑量的百草枯,但每年仍有百草枯中毒死亡的案例發生。有研究[3-4]表明百草枯在血液和尿液中的濃度和中毒時間的相關性是評價患者預后診斷的重要指標,因此,亟待建立快速準確測定百草枯在中毒患者體內濃度的分析方法。
近年來,血液和尿液中百草枯含量測定常用的方法主要有分光光度法[5-6]、氣相色譜法[7](GC)、氣相色譜-質譜聯用法(GC-MS)[8]、液相色譜法(HPLC)[9]、液相色譜-質譜聯用法(LC-MS)[10-11]和毛細管電泳法(CE)[12]等,但這些方法存在諸如耗時長,易受基質效應影響,樣品預處理過程復雜等局限性。相比之下,基質輔助激光解吸電離-傅里葉變換離子回旋共振質譜法[13-14](MALDI-FTICR-MS)具有超高分辨率、靈敏度與精確度高、樣品用量少、制備方便、耗時短、易操作、耐鹽度好等優點,適用于復雜生物樣品中痕量成分的直接快速測定。基于此,本研究建立了一種利用MALDI-FTICR-MS測定全血及尿液中百草枯的方法,以實現對微量生物樣本中百草枯的快速定量分析,方法前處理簡單快速,試劑消耗少,可滿足醫院臨床監測及司法鑒定需求。
1 實驗部分
1.1 儀器與試劑
配有基質輔助激光解吸電離源的Solarix XR 7.0 T傅里葉變換離子回旋共振質譜儀(MALDI-FTICR-MS,德國Bruker公司),配Smartbeam Ⅱ型固態激光器,最大能量300 μJ,頻率在0~1 kHz內可調,波長為355 nm;DataAnalysis 5.0數據處理軟件(德國Bruker公司);BSA224S型電子天平(賽多利斯科學儀器有限公司);CT15RE型離心機(日立工機株式會社);JP-060S型超聲波清洗機(深圳市潔盟清洗設備有限公司);VORTEX 1型旋渦混合器(德國IKA公司)。
百草枯和百草枯-D8標準品(純度分別為82.4%和95.9%,德國Dr.Ehrenstorfer公司);2,5-二羥基苯甲酸(2,5-DHB,德國Bruker 公司);α-氰基-4-羥基肉桂酸(CHCA)、芥子酸(SA)(美國Sigma-Aldrich公司);乙腈(色譜純,美國Honeywell公司);甲酸(LC-MS級,美國Thermo Fisher Scientific公司);三氟乙酸鈉校準溶液(NaTFA,美國Sigma-Aldrich公司);實驗用水為超純水,由Milli-Q超純水系統制備;實際樣品均為客戶委托送檢樣品。
1.2 標準溶液的配制
精密稱取適量(精確至0.000 1 g)百草枯和百草枯-D8標準品,用水溶解并定容,分別配成質量濃度為100 mg/L的標準儲備液,于塑料瓶中-20 ℃冷凍保存。精密移取百草枯標準儲備液適量,用含1%甲酸的乙腈溶液稀釋成質量濃度為5、10、25、50、100、500、1 000、2 000 μg/L的系列標準工作溶液;百草枯-D8儲備液用含1%甲酸的乙腈溶液稀釋成質量濃度為100 μg/L的內標工作溶液。
1.3 樣品制備
1.3.1 提 取取10 μL全血或尿液置于1.5 mL EP(艾本德,Eppendorf)離心管中,其中全血加入25 μL內標工作溶液(100 μg/L)和25 μL含1%甲酸的乙腈溶液,尿液中加入20 μL內標工作溶液(100 μg/L)和20 μL含1%甲酸的乙腈溶液,然后各超聲2 min,再渦旋1 min,最后在室溫下以10 000 r/min高速離心10 min,取上清液,備用。
1.3.2 點 樣精密移取10 μL上清液與10 μL 2,5-DHB(10 g/L,用30%乙腈水溶液配制)溶液等比例混勻,取3 μL混合液點樣于MALDI靶板上,待基質自然晾干形成均勻結晶層后方可進樣分析。
1.4 標準曲線繪制
分別以10 μL空白全血、空白尿液和純水為基質繪制3條標準曲線,配制方法:向空白全血中加25 μL內標(100 μg/L)和25 μL系列標準工作溶液,尿液和純水中分別加20 μL內標(100 μg/L)和20 μL系列標準工作溶液,然后超聲2 min,再渦旋1 min,最后在室溫下以10 000 r/min高速離心10 min。采用“1.3.2”點樣方法進樣分析。
1.5 質譜條件
在正離子模式下采用MALDI源,質量采集范圍為m/z50~300,采樣大小為2 M(兆),累加次數為8;激光能量為30%,頻率100 Hz,激光點數為100 Shots,光斑為Medium,激光路徑為Random。數據采集前,在ESI正/負離子源模式下用0.01 g/L三氟乙酸鈉溶液(用50%乙腈水溶液配制)進行儀器校準;數據采集過程中鎖定內標離子m/z194.165 36進行實時校正,以保證數據采集中質量軸的準確性。定性離子對為m/z186.115 15→171.091 67,定量離子為m/z186.115 15,并采用內標法定量分析(以定量離子峰m/z186.115 15與內標離子峰m/z194.165 36對應的峰強度比值計算)。
2 結果與討論
2.1 MALDI基質的選擇
按“1.3”方法進行樣品提取及點樣,分別在50 μL空白全血、空白尿液和純水中加入100 μL百草枯標準溶液(100 μg/L)和100 μL含1%甲酸的乙腈溶液,考察不同樣品與2,5-DHB、CHCA和SA(濃度均為10 g/L) 3種常用的MALDI基質[15]混合點樣后的分析結果。結果顯示:在相同加標濃度下,全血、尿液和純水樣品用2,5-DHB為基質輔助電離得到的百草枯響應強度最高,故選擇2,5-DHB為MALDI基質輔助百草枯的電離。
2.2 提取條件的優化
由于全血和尿液中均含有大量蛋白質和無機鹽,若直接進質譜會影響百草枯的離子化效率,因此需通過前處理去除干擾成分。本研究采用液液萃取方式提取全血和尿液中的百草枯,方法簡單快速,節省前處理時間。文獻報道采用液液萃取方式提取血液和尿液中百草枯的優選溶劑包括純乙腈[10,16]、含1%甲酸的乙腈溶液[9]和80%乙腈水溶液[17],因此實驗考察了這3種萃取溶劑對全血和尿液中百草枯(空白樣品加標濃度為200 μg/L)的提取效率。結果顯示,采用含1%甲酸的乙腈溶液對全血和尿液中百草枯的提取效率相對較高,且由于百草枯在酸性和中性溶液中相對穩定,而在堿性溶液中易分解[18],考慮到部分尿液可能為堿性,因此選擇含1%甲酸的乙腈溶液為樣品提取溶劑,以使樣品的穩定性更好。
2.3 MALDI條件的優化
MALDI源可優化的條件通常有累加次數、激光能量、激光頻率、激光點數、光斑大小,其中累加次數對百草枯離子峰強度的影響最大。一般以2n增加,8次以內累加次數越多,峰強度越大,但超過8次后峰強度無明顯增加,因此選擇累加次數為8次,即激光在手動選取的中心點附近按照一定規律選取8個位置完成一次采集。增加激光能量、激光頻率、激光點數均會增強離子流且獲得高信噪比的質譜圖,但離子流太強,過剩的離子在和諧阱中將受電荷排斥導致質量軸偏移[19],使最終得到的質譜峰分叉而影響定性和定量分析。激光能量、激光頻率、激光點數和光斑大小分別通過單因素優化,比較對應的峰強度和穩定性,最終確定上述參數優化后分別為30%、100 Hz、100 Shots和Medium光斑模式。
2.4 方法學驗證
2.4.1 專屬性與基質效應的評價分別取3份空白全血和尿液按照“1.3”方法將其中內標溶液換成含1%甲酸的乙腈溶液進行前處理和點樣,在優化條件下測定,結果見圖1。由圖可見,空白全血和尿液在百草枯(m/z186.115 15)和內標(m/z194.165 36)標準溶液對應的離子峰位置并無相應的離子峰出現,雖然附近有其他峰,但FTICR-MS具有超高分辨率,可以很好的將其分開,因此專屬性符合要求。






圖1 百草枯和內標標準溶液(A)、空白全血(B)及空白尿液(C)的質譜圖Fig.1 Mass spectrograms of paraguat and internal standard solution(A),blank whole blood(B) and blank urine(C)
生物樣本在進行質譜分析時,由于本底成分復雜,會對待測組分的離子化產生影響,產生特定的離子增強或抑制作用,這種影響被稱為基質效應(Matrix effect,ME)[20]。本實驗采用標準曲線斜率的比值來評價基質效應,其計算公式為ME=Ka/Kb,其中Ka為樣品基質校準曲線的斜率,Kb為溶劑校準曲線的斜率;ME為100%時表示無基質效應,80%~120%時為弱基質效應,50%~80%或120%~150%時為中等程度基質效應,ME<50%或ME>150%為強基質效應[20]。由表1可見,在全血和尿液的ME值分別為101%和105%,表明此兩種樣本分析痕量百草枯時會產生一定的基質效應,但影響不大。而比較同一百草枯加標濃度下待測物與內標的峰強度和比值,發現全血和尿液對待測物和內標同時產生明顯的離子抑制作用,由于本實驗采用內標法定量,在定量過程中加入內標校正后可消除基質效應的影響,因此可直接采用純溶劑配制標準曲線來進行實際樣品的測定。
2.4.2 線性關系、檢出限與定量下限以質量濃度為橫坐標(x,μg/L),對應的待測物定量離子與內標離子峰強度的比值為縱坐標(y)繪制標準曲線。結果顯示,全血、尿液、水3種基質配制的標準溶液在10~5 000 μg/L質量濃度范圍內線性良好,相關系數(r)為0.995 5~0.999 2(表1)。以3倍信噪比(S/N=3)計算得全血、尿液和純水中百草枯的檢出限(LOD)分別為3.0、1.5、0.6 μg/L,定量下限(LOQ,S/N=10)分別為10、5.0、2.0 μg/L。

表1 百草枯在全血、尿液及水中的線性方程、線性范圍、相關系數、基質效應、平均回收率、相對標準偏差(RSD)、檢出限及定量下限Table 1 Linear equations,linear ranges,correlation coefficients,matrix effects,average recoveries,RSDs,LODs and LOQs of paraquat in whole blood,urine and water
2.4.3 準確度與精密度分別取空白全血和尿液,進行低、中、高3個濃度水平的加標回收實驗,按“1.3”方法進行樣品制備,每個濃度水平平行制備6個樣品,在優化條件下測定,計算方法的回收率和相對標準偏差(RSD)。結果顯示,百草枯在全血和尿液2種生物樣品中3個加標濃度下的回收率為85.2%~110%,RSD為0.40~7.3%(表1),表明方法的準確度和精密度良好,可滿足檢測要求。
2.4.4 穩定性評價采用空白樣品加標分別考察了全血和尿液中百草枯在室溫下24 h內、反復凍融3次(-20 ℃)、在-20 ℃儲存條件下7 d和30 d的穩定性實驗,每個條件平行3次。結果顯示,百草枯在全血和尿液中的相對標準偏差為0.5~7.7%,回收率為90.1%~106%,說明本研究所建立分析方法的穩定性符合要求,樣品在-20 ℃儲存條件下30 d內穩定。
2.4.5 與其他方法的比較將本方法與已報道的生物樣本中百草枯的檢測方法進行比較(表2),由表中數據可見,本方法所需樣品量和溶劑量更少,且百草枯的檢出限更低。此外,本法采用液液萃取法對樣品進行前處理,只需一步萃取即可上樣分析,分析時間僅十幾秒,顯著縮短了檢測時間,可滿足臨床及司法鑒定中百草枯中毒快速檢測的需求。
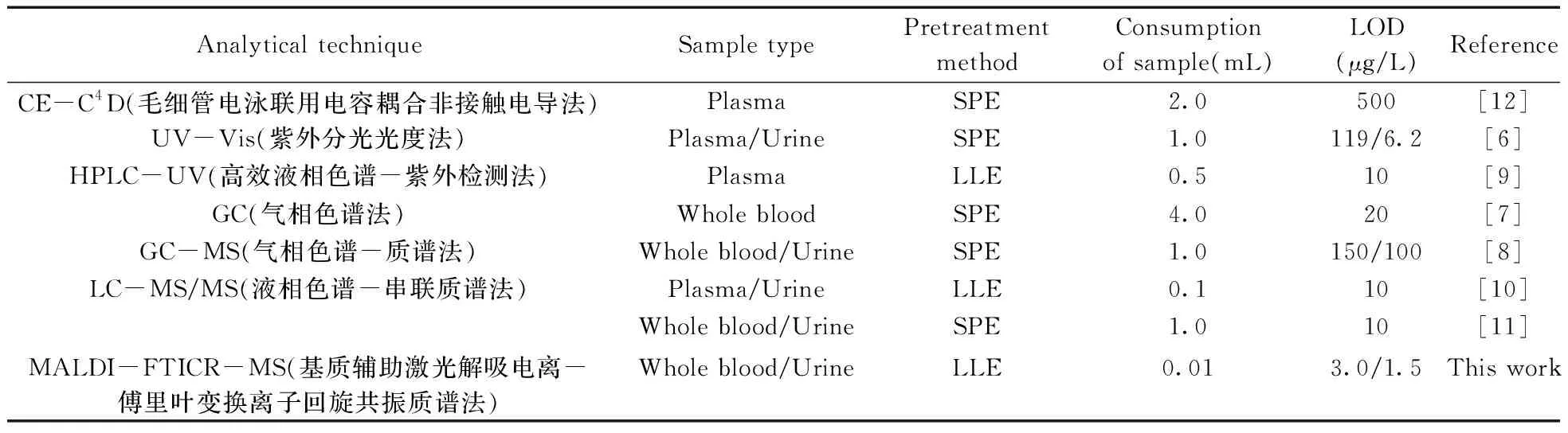
表2 與其他文獻報道的生物樣本中百草枯檢測方法的比較Table 2 Comparison with the reported analytical techniques for the determination of paraquat in biological samples
2.5 實際樣品的測定
采用本方法對3份來自醫院百草枯中毒病人的全血和尿液進行檢測。結果顯示,3份病人全血中分別檢出百草枯含量為0.794、5.45、0.049 4 μg/L,RSD分別為5.7%、8.0%、5.4%;對應病人的尿液中百草枯檢出含量分別為2.58、153、0.818 μg/L,RSD分別為1.5%、1.4%、3.7%。3份病人尿液中的百草枯含量明顯高于血液,表明病人中毒后,大量百草枯可通過尿液排出體外。其中1份病人的全血和尿液樣品的質譜圖見圖2,該病人全血及尿液中所檢出的百草枯含量分別為0.049 4、0.818 μg/L。

3 結 論
本研究建立了一種利用MALDI-FTICR-MS測定全血及尿液中百草枯的方法,以含1%甲酸的乙腈溶液為提取溶劑,采用液液萃取法從生物樣品中提取百草枯,提取液與2,5-DHB(10 g/L)基質混合后即可點樣分析。該方法具有樣品和溶劑用量少、操作簡單、分析速度快和靈敏度高等優點,可滿足臨床及司法鑒定中百草枯中毒快速檢測的需求。
