通過式高效凈化/超高效液相色譜-串聯質譜法測定化妝品中73種糖皮質激素
2020-09-29 03:51:16楊飄飄李麗霞
分析測試學報 2020年9期
劉 紅,楊飄飄,李麗霞
(1.湖北省藥品監督檢驗研究院,湖北 武漢 430075;2.湖北省藥品質量檢測與控制工程技術研究中心,湖北 武漢 430075)
糖皮質激素類藥物由Kendall于1935年首次發現,20世紀50年代以來,許多腎上腺糖皮質激素類藥物被合成,其核心結構為環戊烷多氫菲核[1]。該類藥物可抑制纖維細胞增生,減少5-羥色胺形成,因而對皮膚有一定的嫩白作用[2]。為謀取高額利潤,一些不法商家在化妝品中違法添加糖皮質激素,然而長期濫用糖皮質激素可引起激素依懶性皮炎[3],臨床表現為皮膚明顯變薄、毛細管擴張、痤瘡和色素沉著等癥狀。《化妝品安全技術規范》(2015年版)規定糖皮質激素為禁用組分[4],涉及到49種糖皮質激素;GB/T 24800.2-2009涵蓋41種糖皮質激素[5]。但為規避監管,仍有不法商家非法添加標準方法之外的糖皮質激素[6]。因此,研究出一種同時測定化妝品中更多種糖皮質激素、更易操作、準確定性定量的檢測方法尤為重要。
目前糖皮質激素類藥物的檢測手段主要有薄層色譜法[5]、高效液相色譜法[7-9]、高效液相色譜-串聯三重四極桿質譜法[10-13]和高效液相色譜-高分辨質譜法[14-15]等,但薄層色譜法常用于定性篩查,無法準確定量;高效液相色譜-串聯三重四極桿質譜法在定量方面的靈敏度優于高效液相色譜法和高效液相色譜-高分辨質譜法,故應用最為普遍。由于化妝品基質復雜,目標物結構相似,目前報道的前處理方法有固相萃取法[5,16-17]或QuEChERS法[18],但這些方法具有步驟繁瑣、耗時等不足。
本研究針對化妝品基質復雜、以油性原料和水為主的特性,采用通過式高效凈化除去脂溶性物質,凈化效率高,操作簡便,結合超高效液相色譜-串聯質譜(UPLC-MS/MS)法對水劑、乳液、膏霜3類基質化妝品中73種糖皮質激素進行了測定。方法快速、簡單、穩定,結果可靠,能夠滿足化妝品中糖皮質激素風險篩查及監測工作的需要。
1 實驗部分
1.1 儀器、試劑與材料
Waters Acquity UPLC/XEVOTMTQ-S 液相色譜-三重四極桿串聯質譜儀(美國 Waters 公司);XP504電子天平(瑞士梅特勒公司);Milli-Q 超純水器(美國Millipore 公司);LC-250超聲波清洗機(山東濟寧魯超超聲設備有限公司);臺式離心機(德國Thermo Biofuge公司)。Waters CORTECS C18(150 mm×2.1 mm,2.7 μm,美國 Waters 公司);固相萃取柱為Oasis?PRiME HLB(200 mg/6 mL,60 mg/3 mL,美國Waters公司);甲醇、乙腈(色譜純,默克股份兩合公司);甲酸銨、甲酸(色譜純,Aladdin 公司);乙酸銨(色譜純,Macklin公司)。
73種糖皮質激素標準物質(見表1)分別購自中國食品藥品檢定研究院、歐洲藥品質量管理局、美國藥典委員會、加拿大 TRC 公司、美國Stanford 分析化學公司、德國 Dr.Ehrenstorfer 公司、美國A ChemTek公司。
1.2 對照溶液的配制
精密稱取各對照品適量置于10 mL容量瓶中,加甲醇溶解并稀釋至刻度,搖勻,即得100 μg/mL的各單標對照儲備溶液。分別精密量取適量各單標對照儲備溶液于同一10 mL容量瓶中,加甲醇稀釋至刻度,搖勻,即得200 ng/mL的混合標準儲備液。
1.3 樣品制備
稱取均勻試樣約0.2 g,置于15 mL離心管中,加入飽和氯化鈉溶液3 mL,渦旋30 s,分散均勻,精密加入4 mL乙腈,渦旋30 s,超聲提取30 min,渦旋混合搖勻,取上清液過PRiME HLB柱,重力作用下收集全部流出液。經0.22 μm濾膜過濾,濾液供UPLC-MS/MS測定。
1.4 色譜條件
色譜柱:Waters Cortecs C18(150 mm×2.1 mm,2.7 μm);柱溫:35 ℃;流速:0.3 mL·min-1;進樣體積:2 μL;流動相:乙腈(A)和0.1%乙酸溶液(B)。梯度洗脫程序:0~6 min,5%~15% A;6~16.8 min,15%~23.4% A;16.8~18.8 min,23.4% A;18.8~38 min,23.4%~42% A;38~44 min,42%~67.8% A;44~46 min,67.8% A;46~50 min,67.8%~85% A;50~52 min,85%~5% A;52~56 min,5% A。
1.5 質譜條件
離子源:電噴霧離子源;掃描方式:正離子掃描;檢測方式:多反應監測(MRM);去溶劑氣流速:800 L/h;去溶劑溫度:350 ℃;毛細管電壓:3.2 kV;73種待測物的質譜參數見表1。
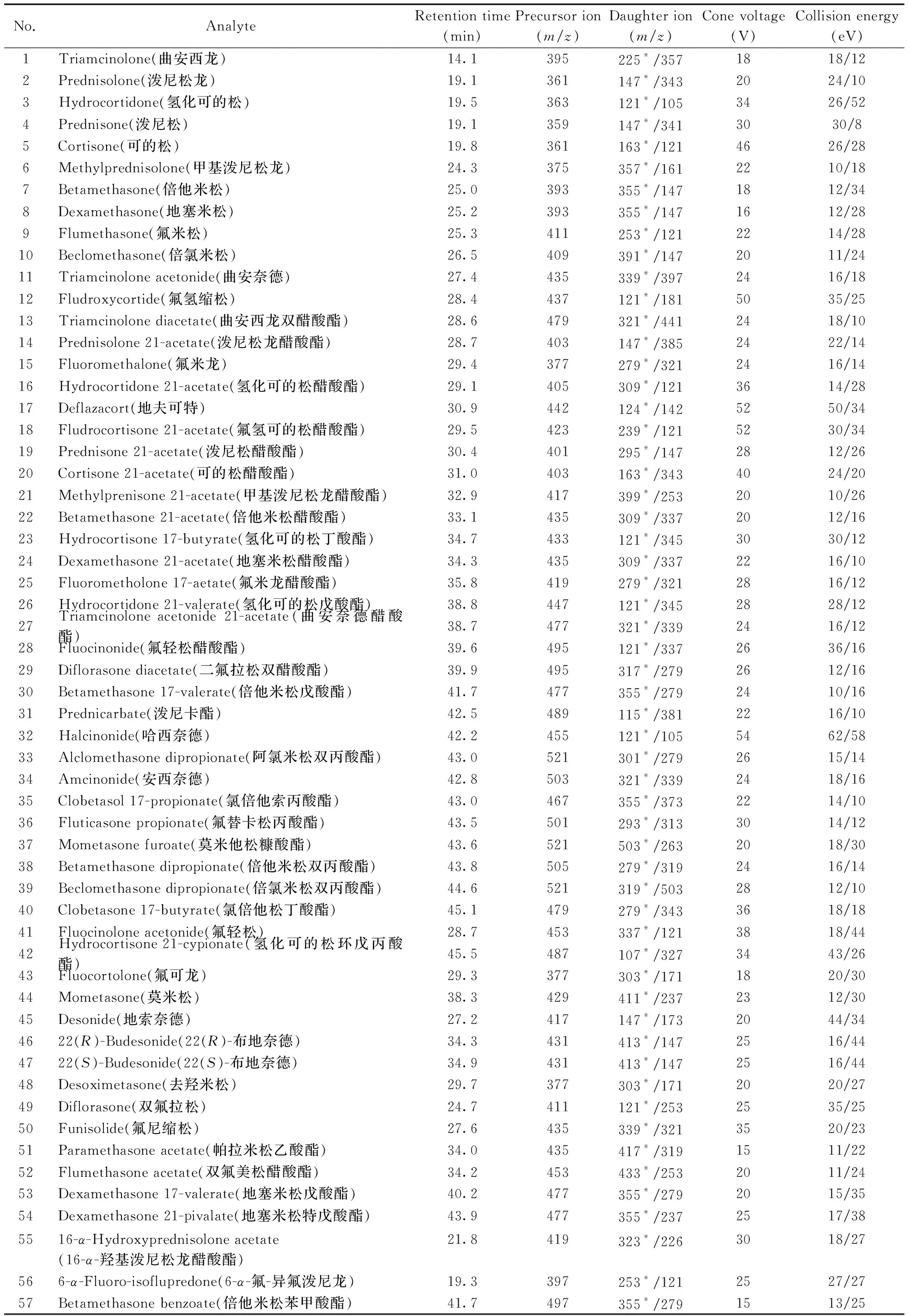
表1 73種分析物的質譜參數Table 1 MS parameters of the 73 analytes
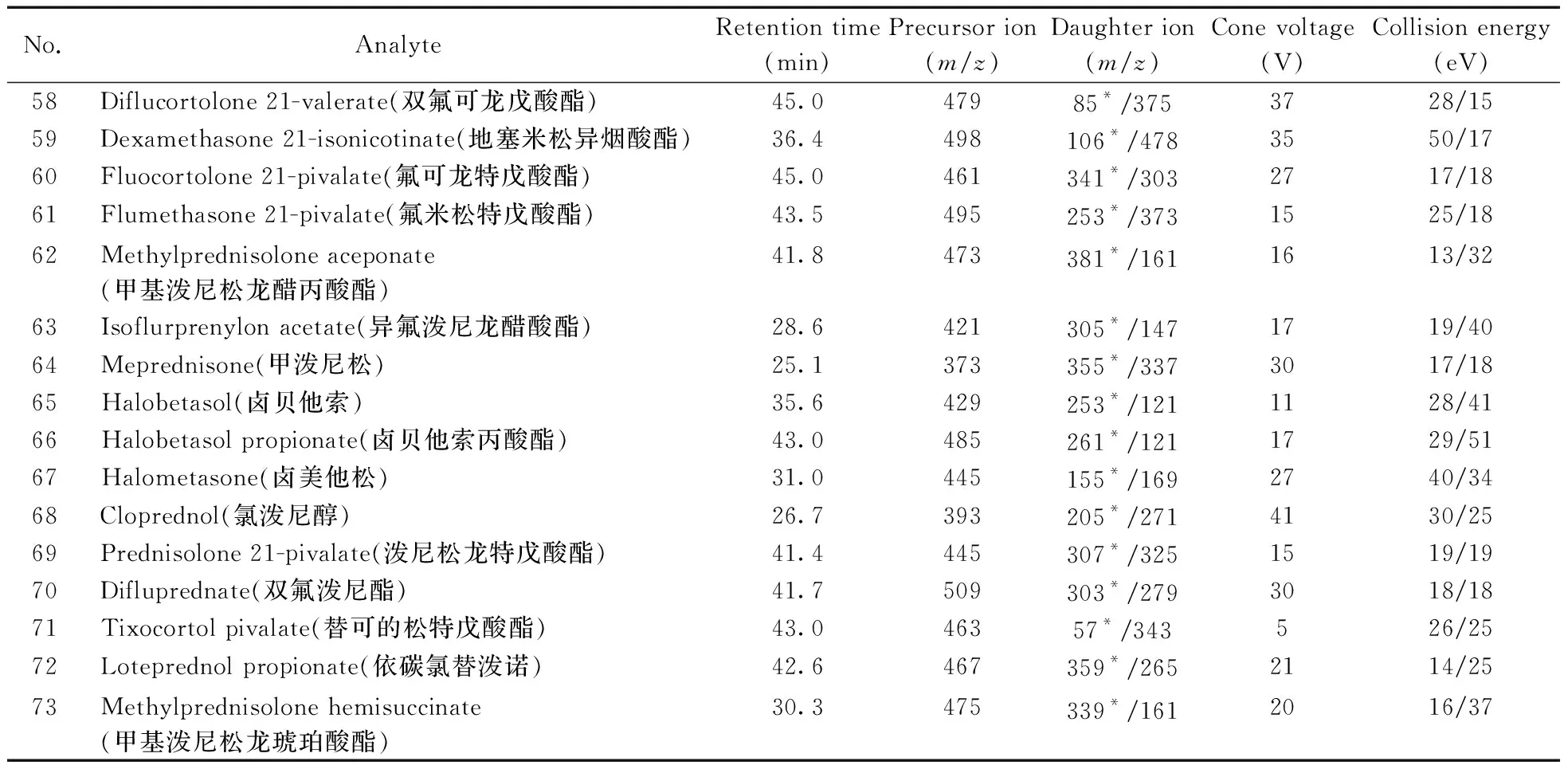
(續表1)
2 結果與討論
2.1 樣品提取方法的優化
糖皮質激素為甾體化合物,其結構特征是在固醇核D環的C17上有α羥基,在C環的C11上有氧或羥基[19]。因此,糖皮質激素類化合物多屬于弱極性或中等極性,文獻[16-17]以乙腈或甲醇為提取溶劑。考慮到乙腈作為提取溶劑更易分層,不易乳化,故選用乙腈為提取溶劑。化妝品基質復雜,由油性、粉質、溶劑類、膠質、表面活性劑、香精香料、顏料色素、保濕劑、防曬劑、防腐劑和抗氧化劑等原料根據不同的產品需求組合而成[20],所以分散劑的選擇至關重要。實驗比較了以水、飽和氯化鈉為分散劑時73種糖皮質激素的回收率。結果表明,飽和氯化鈉作為分散劑時,有73%(53/73)的目標物回收率更接近100%,而水作為分散劑時,僅有27%(20/73)的目標物回收率更接近100%,分析原因可能為飽和氯化鈉屬強電解質,作為分散劑和破乳劑可增加樣品的表面張力,使分散更完全。所以最終選擇飽和氯化鈉作為分散劑。
2.2 色譜條件的優化
糖皮質激素以環戊烷多氫菲核為母核,僅取代基位置不同,本研究中共有12組同分異構體,如22(R)-布地奈德和22(S)-布地奈德等。采用0.1%乙酸溶液和乙腈為流動相,比較了Agilent Poroshell EC-C18(100 mm×2.1 mm,2.7 μm) 、Waters Cortecs C18(150 mm×2.1 mm,2.7 μm)和Waters BEH C18(100 mm×2.1 mm,1.7 μm)3種色譜柱對目標化合物分離效果和峰形的影響。結果表明,73種藥物在3種色譜柱上的峰形均較好,但分離效果差異較大,12組同分異構體在Agilent Poroshell EC-C18(100 mm×2.1 mm,2.7 μm) 和Waters BEH C18(100 mm×2.1 mm,1.7 μm)上不能同時分離,而在Waters Cortecs C18(150 mm×2.1 mm,2.7 μm)上能實現較好分離,因此本文選用Cortecs C18(150 mm×2.1 mm,2.7 μm)色譜柱。
考察了乙腈-0.1%乙酸溶液、乙腈-0.1%甲酸溶液、乙腈-0.01 mol·L-1甲酸銨和乙腈-0.01 mol·L-1乙酸銨4種流動相體系對目標化合物分離效果和信號強度的影響。結果顯示,上述4種流動相體系下,73種糖皮質激素的分離度和保留時間相似,但以乙腈-0.1%乙酸溶液作為流動相時的峰形最好,且絕大部分目標化合物的響應優于其他流動相體系。故選擇乙腈-0.1%乙酸溶液作為流動相。

圖1 典型同分異構體的基峰色譜圖Fig.1 Base peak chromatogram of typical isomersthe peak numbers denoted were the same as those in Table 1
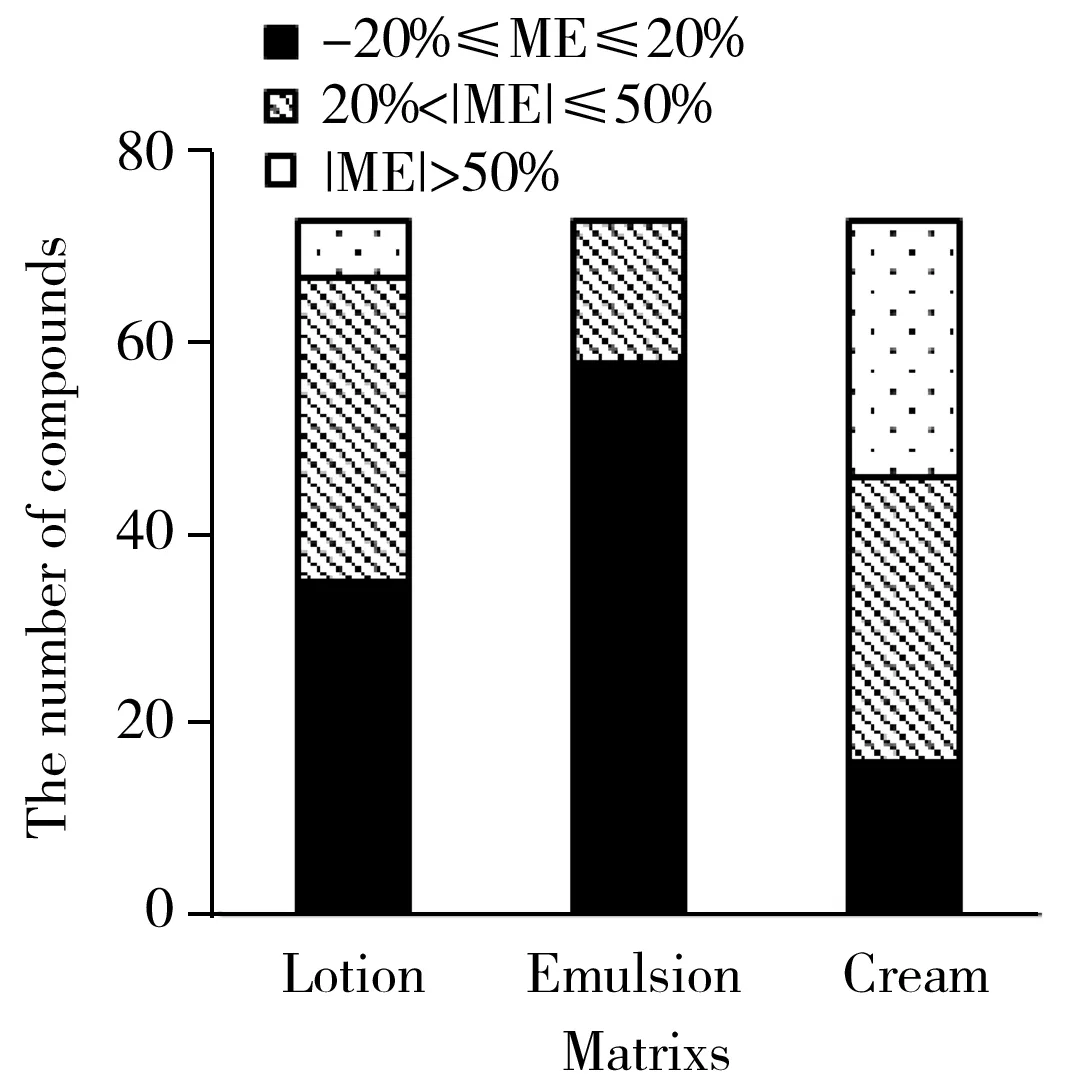
圖2 3種基質化妝品基質效應的比較Fig.2 The comparison of matrix effect for three matrices
2.3 質譜條件的優化
在電噴霧離子源下,分別對質量濃度為200 ng/mL的目標化合物對照溶液做正離子和負離子全掃描,發現73種目標化合物均在正離子模式下獲得響應最佳的準分子離子峰。由于糖皮質激素有相同的母核,73種目標化合物中有12組同分異構體,而且有些化合物雖不是同分異構體,但其質量數相同,裂解規律相似,易產生相同的碎片,所以選擇特征的、無干擾且穩定的子離子對作為定量定性離子,以多反應監測模式優化各質譜參數,使化合物的準分子離子與特征碎片離子產生的離子對強度達到最大。優化條件下典型同分異構體的色譜圖見圖1。
2.4 凈化條件的優化
樣品經乙腈萃取后的溶液中含有大量脂溶性物質[21],因此利用固相萃取柱凈化樣品溶液。有文獻采用HLB固相萃取柱凈化[5,16-17],但步驟繁瑣、耗時較長。而新型PRiME HLB 柱屬于過濾型固相萃取柱,無需活化和平衡,能有效吸附脂溶性干擾物,過程簡單、高效。因此,本實驗選取200 mg/6 mL和60 mg/3 mL兩種型號的PRiME HLB固相萃取柱進行了比較。結果顯示,與過柱前相比,凈化后的提取液澄清度均有顯著提升。對兩柱的提取回收率進行統計學t檢驗,結果表明兩者有顯著性差異,60 mg/3 mL固相萃取柱的平均提取回收率更好,故選擇PRiME HLB(60 mg/3 mL)固相萃取柱進行凈化。
2.5 基質效應
基質效應(ME)是由于樣品在離子化時基質成分與目標化合物相互競爭電離所致,包括基質增強效應和基質抑制效應。采用ME=(1-基質對照溶液的峰面積/溶劑對照溶液的峰面積)×100%對73 種化合物的基質效應進行評價。當-20%≤ME≤20%時為弱基質效應;當 20%<|ME|≤50% 時為中等基質效應;當 |ME|>50% 時為強基質效應[22];通常采用基質標準曲線計算以獲得更真實的樣品數據[23]。本研究對水劑、乳液、膏霜3類化妝品的基質效應進行考察,由圖2可知,膏霜類化妝品受基質影響較大,水劑次之,乳液類化妝品受基質效應影響最小。為了能更準確地測定目標化合物,本實驗采用基質匹配標準曲線消除基質效應。
2.6 方法學考察
2.6.1 定量下限、標準曲線、線性范圍在國食藥監許[2010]455號《化妝品中禁用物質和限用物質檢測方法驗證技術規范》中,定量下限定義為能夠對被測物質準確定量的最低濃度或質量[24]。在保證準確定性的前提下,通過使用空白基質逐級稀釋對照品溶液來考察定量下限(LOQ),以信噪比S/N≥10確定73種糖皮質激素的LOQ為0.1~0.3 μg/g(見表2)。
空白樣品中添加系列不同質量濃度的73種混合標準溶液,按“1.3”方法處理得到樣品提取液,以目標化合物的色譜峰面積(y)對其質量濃度(x,ng/mL)進行線性回歸,得到線性方程和相關系數。結果表明,73種目標化合物在各自質量濃度范圍內線性關系良好,相關系數均大于0.99(見表2)。
2.6.2 回收率與相對標準偏差選取水劑、膏霜、乳液3類空白基質,按1、2、5倍LOQ進行加標回收實驗,每個加標水平平行分析3次。實驗數據顯示,水劑的平均回收率為88.1 %~118%,相對標準偏差(RSD) 為 0.40%~14%;膏霜的平均回收率為73.3%~119%,RSD為 0.40%~18%;乳液的平均回收率為86.5%~115%,RSD為 0.60%~17%。典型乳液基質的數據見表2。

表2 73種化合物在典型乳液基質中的線性范圍、相關系數、回收率、精密度和定量下限Table 2 Linear ranges,correlation coefficients,average recoveries,precisions and limits of quantitation of the 73 compounds in typical emulsion matrices
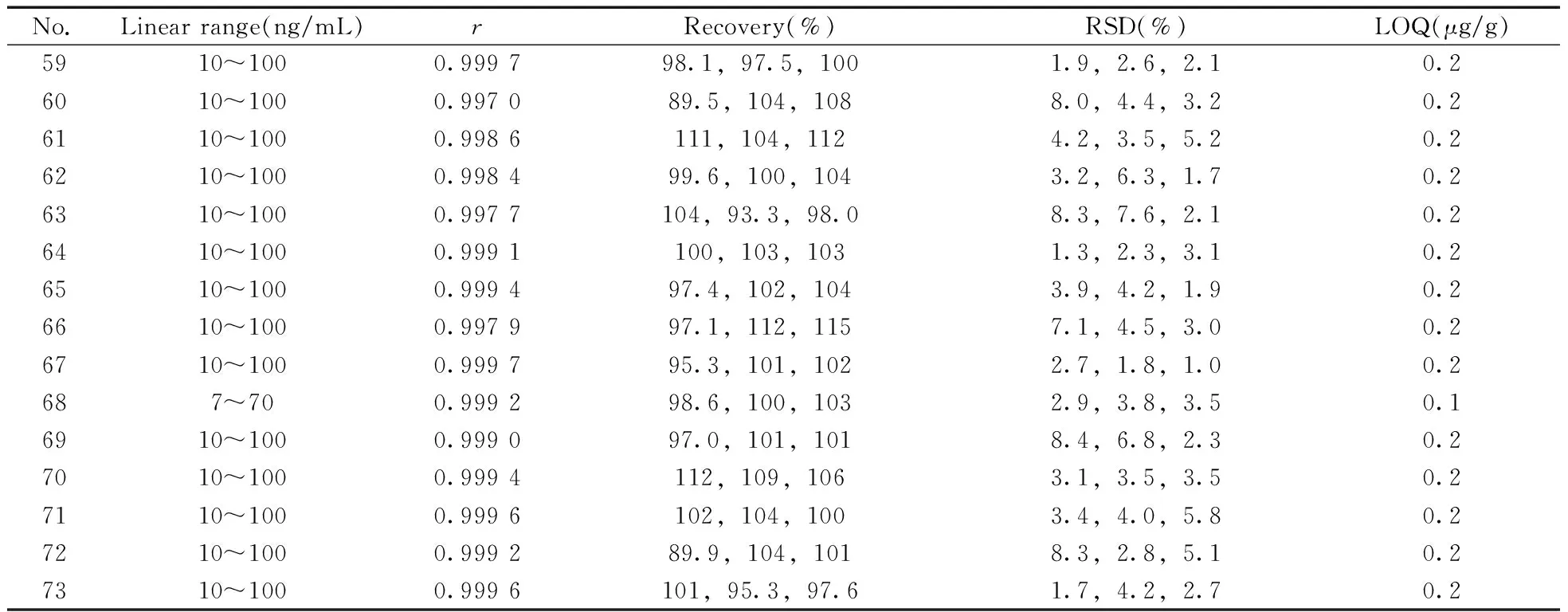
(續表2)
2.7 實際樣品測定
采用本方法對客戶委托的7個樣品進行了檢測,其中2批樣品檢出倍他米松,含量分別為4.9 μg/g和5.2 μg/g。典型陽性樣品的總離子流圖及提取離子流圖見圖3。
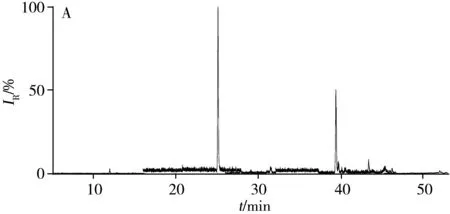
3 結 論
本研究建立了通過式高效凈化/超高效液相色譜-串聯質譜檢測化妝品中73種糖皮質激素的分析方法。該方法樣品前處理簡單,定量準確,靈敏度高,能滿足同時測定化妝品中73種糖皮質激素的要求,為有效監管化妝品中非法添加禁用物質提供了技術支撐。
