陽離子交換皂石黏土在基質輔助激光解吸電離飛行時間質譜分析中的應用
2020-08-17 07:55:14丁宇琦朱價周霞羅金文
浙江大學學報(理學版) 2020年4期
關鍵詞:分析
丁宇琦,朱價,周霞,羅金文
(浙江省食品藥品檢驗研究院,浙江杭州310012)
糖類化合物是自然界中分布很廣的一類重要化合物,在生命體的代謝組學和營養學等中扮演著極其重要的角色,是一切生命體維持生命活動所需能量的最主要來源[1]。 然而,持續的高血糖會導致多尿、多飲、多食和體重減輕等,最終造成各身體組織的慢性損害和功能障礙(糖尿病患者的主要癥狀)。 目前我國對糖尿病的診斷依據是1999 年世界衛生組織頒布的標準,主要根據空腹和餐后的血糖量進行綜合判定。 因此,快速準確地測定血液和血漿中的糖類化合物(尤其是單糖)含量,對于精確診斷糖尿病有重要意義。
糖類化合物難揮發、熱不穩定,其中多糖還具有相對分子質量分布發散的性質,其質譜表征比較困難。為此,科研工作者做了大量研究[2],現有文獻報道的糖類檢測技術主要有:高效液相色譜法(HPLC)、離子色譜法(IC)、薄層色譜法(TLC)以及氣相色譜-質譜聯用法(GC-MS)等。例如LIU 等[3]利用親水液相色譜技術分離了地黃中的9 種糖類化合物;ZHANG 等[4]用苯硼酸解吸電噴霧離子化法檢測糖類;FUZFAI 等[5]將氣相色譜串聯離子阱質譜技術用于各種糖類中的三甲基硅烷基及其肟衍生物的特征裂解規律分析。 以上技術雖然比較成熟,但均存在不足,例如:氣相色譜法需要對不易揮發的糖類進行衍生化處理,費時費力;薄層色譜法分離效果差,操作時間長,難以準確定量;液相色譜法多采用示差折光檢測器,靈敏度低,易受環境影響,重復性差;而離子色譜法則需要高性能的陰離子交換柱。
MALDI-TOF-MS 是一種新型的軟電離技術,具有許多優點。首先,檢測結果幾乎不受污染物和雜質影響,由于離子化效率很高,可分析一些較難電離的樣品,得到完整的電離產物;其次,樣品前處理方法非常簡單,甚至可以直接分析未處理的生物樣品,儀器操作也很容易。 相較傳統檢測技術,MALDI-TOF-MS 技術具有分辨率高、操作簡便、快速準確、質量分析范圍大、能夠直接分析非衍生化物質等優點,并可測定精確分子量,定性準確度高。 因此,該技術已被廣泛應用于多肽、聚合物、聚核苷酸等生物大分子物質分子量的測定及蛋白質高通量的鑒定。 但將該技術應用于糖類的研究報道較少,且主要集中于糖蛋白中的糖鏈分析、大分子多糖精確分子量的測定等[6-8],對小分子糖類化合物的研究尚處于探索階段。 PARK 等[9]用常規有機基質二羥基苯甲酸(DHBA)及MALDI-TOF-MS 技術分析了啤酒中的寡糖,結果表明,在溶劑中添加陽離子化試劑有利于糖類的離子化,但未對離子化過程和基質的工作原理進行探討;NIMPTSCH 等[10]同樣將 DHBA 作為基質,利用MALDI-TOF-MS 技術進行了黏多糖和低聚糖硫酸鹽的檢測;劉悅玫等[11]利用MALDI-TOF-MS 技術進行了復雜糖一致性研究;裴興麗等[12]以DHBA 為基質,利用MALDI-TOF-MS 技術對大豆食品中寡糖成分進行了成像分析。這些文獻僅提出寡糖類化合物的檢測方法,未涉及小分子糖類化合物,其主要原因是在解吸過程中同時發生基質本身的離子化和碎裂,對圖譜的解析困難較大,難以判斷某個峰是來源于基質還是分析物,從而大大限制了MALDI-TOF-MS技術在小分子糖類化合物領域的應用,特別是針對未知化合物的分析。 因此,引入無基質的激光解吸電離技術(matrix-free techniques)[13]、對傳統有機基質進行修飾(例如通過加入基質添加物形成復合基質)[14]、制備新型無機基質(例如 Au,Ag 等納米材料)[15]等成為攻克上述難題的關鍵。
由于硅酸鹽層之間的相互作用力較弱,皂石黏土很容易在水中膨脹形成溶凝膠,另外,黏土還有較強的陽離子交換和分子嵌入能力,在多個領域已有廣泛應用[16-17]。 在化學領域主要作為催化劑[18]以及納米復合材料的載體[19],在MALDI-TOF-MS 技術中尚未見應用。 本文選取一種人工合成的蒙脫石皂石黏土,并將有機基質THAP 成功嵌入經陽離子交換后的皂石黏土的晶面間隙中,形成復合基質,用于糖類化合物的分析。結果表明,由于受該復合基質晶面間距的限制,僅能使部分小分子糖類化合物離子化,具有小分子選擇性。方法操作簡便、分析效率高、經濟成本低。
1 試驗部分
1.1 儀器設備和試劑
MALDI-TOF-MS 配有 337 nm 波長的氮氣激光(Waters 公司);黏土(Sumecton SA)由日本Kunimine 材料實業公司提供;2,4,6-三羥基苯乙酮(THAP,168 Da)、多肽物質 P(SubP)購自德國CNW 公司;葡萄糖(D-glucose,Glu)、半乳糖(D-galactosel)、纖維二糖(D-cellobiose)、棉子糖(D-raffinose)、麥 芽 六 糖(Maltohexaose)、麥 芽 七 糖(Maltoheptaose)、環糊精(γ-cyclodextrin)均購自上海安譜實驗科技股份有限公司;乙腈(色譜純)購自Merk 公司;試驗用水為Milli-Q 超純水。
1.2 儀器參數
激光能量約8 μJ,激光射速10 Hz,正離子反射模式下加速電壓17 kV,延遲時間500 ns。
1.3 分析物前處理
Na+交換的黏土(NaSm):將 0.1 mol CH3COONa 溶于50 mL 水中,待完全溶解后與500 mg 黏土混合形成懸濁液,在70℃下攪拌1 h,用0.2 μm 濾紙過濾,去除溶液;在沉淀物中再加入相同體積和濃度的CH3COONa 溶液,重復反應3 次,將最終的沉淀物冷凍干燥過夜。
復合基質:分別稱取4 mg THAP 基質和一定量的 NaSm(THAP 和 NaSm 的質量比分別為4∶0.25,4∶0.5,4∶1),加入體積比為 7∶3 的乙腈和水溶劑1 mL,保持每份溶液中THAP 的濃度為4 mg·mL-1,超聲溶解 15 min,并稱取 4 mg THAP 按上述方法配成純基質溶液進行對比實驗。
分析物制樣:稱取分析物0.1 mg,用上述溶劑充分溶解并稀釋至濃度為1 μg·mL-1,分別移取相同體積的復合基質溶液和分析物溶液,超聲混合30 min,取2 μL 混合液滴至不銹鋼樣品板上,置于空氣中使溶劑自然揮發,最后將樣品板置入MALDI儀器檢測。
2 結果與討論
2.1 Glu 質譜分析結果
葡萄糖是自然界分布最廣且最為重要的一種單糖,是活細胞的能量來源和新陳代謝的中間產物,是生物的主要供能物質。 由THAP 和NaSm 組成的復合基質(THAP-NaSm)應用于Glu 分析的實驗結果如圖1 所示(縱坐標為優化后的相對強度,各質譜圖相對強度的坐標范圍一致,下同)。 由于Glu 極性較強,在常規質譜條件下質子化非常困難,因此無法觀察到[Glu+H]+峰,而Na+、K+等堿金屬離子與Glu 有較強的結合能力,使得Glu 的離子化峰可以以堿金屬加和峰形式呈現。 圖1(a) 為以THAP 單獨作為有機基質測定的Glu 質譜圖。 由圖1(a)知,僅以THAP 作為基質對葡萄糖進行檢測,基質的Na+加和的離子化峰([THAP+Na]+)并不明顯,更無法觀察分析物的離子化峰(信噪比:S/N=6,R=0.25),說明THAP 并不適合作為葡萄糖的分析基質,而加入NaSm 形成復合基質后,離子化峰有明顯增強。圖1(b)為用復合基質(THAP 和NaSm 的質量比為4∶0.25)后測得的質譜圖,相較圖1(a),分析物的離子化峰([Glu+Na]+信號)強度增強了約35倍(S/N=210,R=0.63),分析物的離子化效率顯著增強,[THAP+Na]+信號的強度增強了約4 倍。當繼續增加 THAP 和 NaSm 的質量比至 4∶0.5 時,[Glu+Na]+的信號強度增強了約100 倍(S/N=590,R=0.83),并且可以清晰地觀察到[Glu-H+2Na]+峰,如圖1(c)所示。當繼續提高THAP 和NaSm 的質量比至4∶0.75 和4∶1 時,絕大部分化合物離子化峰的信號強度出現大幅下降,如圖1(d)和(e)所示,這是因為當混合過多的NaSm 后,激發區的基質濃度被稀釋,從而導致離子化效率降低。 顯然,組成復合基質的THAP 與NaSm 存在一個最優的質量比。
為了確定圖1(b)和(c)中峰信號的增強是由添加NaSm 所致,用 NaCl 取代NaSm,形成質量比為4∶0.5 的 THAP-NaCl 復合基質,再進行葡萄糖質譜分析,結果見圖1(f)。 相較圖1(c),在圖1(f)中未見離子化峰信號增強,因此,推斷NaSm 不僅可以作為高效的Na+源,還可顯著促進分析物和基質的解吸效率。
2.2 多肽物質P(Sub P)的質譜分析結果
多肽物質P 是一種廣泛分布于神經纖維內的神經肽,隨著MALDI 技術的發展,包括THAP 在內的各種常用有機基質已廣泛應用于多肽分析。由于多肽物質P 比糖類物質容易離子化,因此,即使不進行基質修飾,僅以THAP 作為基質仍可觀察到非常明顯的離子化峰,如圖2(a)所示。然而,在混入NaSm形成復合基質后,分析物的質譜峰不僅沒有增強,反而顯著下降,如圖2(b)所示,說明在對分子質量較大的多肽物質進行分析時,復合基質并沒有表現出類似于對糖類化合物分析時的卓越性能。基于以上分析,推測分析物的相對響應值可能與分子大小有關;分子直徑大于THAP-NaSm 晶面間距的化合物無法有效進入復合基質的晶面間隙與分布在晶面內的基質接觸,使得其無法有效離子化,從而導致質譜響應大幅下降。因此,所提出的分析方法對糖類分子具有分子尺寸選擇性。

圖2 以THAP(a)和THAP-NaSm(b)作為基質的多肽物質P 的質譜圖Fig.2 Mass spectra of Sub P measured with THAP(a)and THAP-NaSm(b)as the matrix
2.3 驗證實驗
2.3.1 X 射線衍射(XRD)分析
粉末X 射線衍射(XRD)是評估基質添加物內部晶體結構的重要表征手段,本研究測定了Sm,NaSm 和 THAP-NaSm 的 XRD 圖譜,分別計算了不同添加物的晶面間距,結果如表1 所示。顯然由于THAP 的嵌入,NaSm 的晶面間距變大了。
2.3.2 混合晶體制備方法對比
除了超聲攪拌混合制樣法外,還考察了另外2種混合制樣法,即將THAP-NaSm 與Glu 以機械攪拌的方式以及渦旋振蕩1 h 的方式分別進行混合處理,之后制備樣品板(除混合方式不同外,其余步驟均相同),質譜結果見圖3。對比圖3(a)和圖1(a),兩者所有峰([Glu+Na]+,S/N=12)的響應都基本一致,可知以機械攪拌方式制備的復合基質無任何效果;渦旋振蕩1 h 制備的復合基質在離子化效率上有一定提升,分析物的質譜峰([Glu+Na]+,S/N=180)提升了約15 倍,大致相當于圖1(b)的結果,僅相當于圖1(c)分析物響應的1/3。由于 THAP 主要嵌入NaSm 的晶面間隙內,而機械攪拌和渦旋振蕩方式無法使Glu 有效進入NaSm 的晶面間隙,因此用這2 種方式制備的復合基質離子化效率較低。

表1 Sm,NaSm 和 THAP-NaSm 的 XRD 分析結果Table 1 XRD measurements for Sm,NaSm and THAPNaSm
2.3.3 不同分析物對比
為了驗證對分子尺寸具有選擇性的假設,選擇了 6 種常見的碳水化合物:半乳糖(galactose, 單糖)、纖維二糖(cellobiose, 雙糖)、棉子糖(raffinose,三糖)、麥芽六糖(maltohexaose, 六糖)、麥芽七糖(maltoheptaose, 七糖)、γ-環糊精(cyclodextrin, 八糖)作為分析物,并用THAP-NaSm 作為復合基質
進行分析,質譜結果如圖4 所示。所有圖譜中基質的相關離子化峰的響應幾乎一致,但是分析物相關離子化峰的響應則隨單糖單元聚合數的增多而明顯降低,其中γ-環糊精擁有8 個單糖單元,其相關離子峰僅勉強可見。SUZUKI 等[20]采用THAP 和絲光沸石(LiM20)作為復合基質,成功檢測出了麥芽六糖。 其結果顯示,分析物和基質的相關離子化峰大致相當,說明不同糖類化合物之間的理化性質差異并不是引起圖4 所示的分析物響應下降的主要因素。由圖5 可知,化合物峰離子與基質峰離子的豐度比([M,analyte+Na]+/[matrix+Na]+)隨糖類化合物直徑(根據范德華假設計算)的增大呈指數下降,說明大分子糖類化合物很難進入復合基質的晶面間隙,無法與THAP 充分接觸及相互作用,導致離子化效率降低。因此,糖類化合物的分子直徑大小是導致質譜響應值變化的主要因素。而THAPNaSm 復合基質對小分子糖類化合物的檢測具有較強的選擇性。

圖3 分析物混合晶體的質譜圖Fig.3 Mass spectra of Glu subjected to different analyte/matrix preparation methods
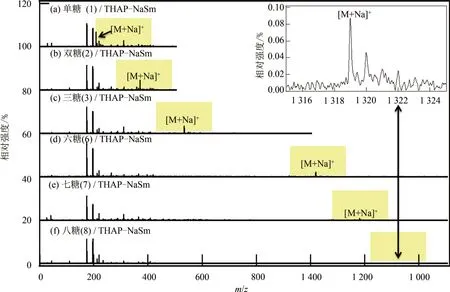
圖4 復合基質測得的糖類化合物質譜圖Fig.4 Mass spectra of various saccharides measured with THAP-NaSm
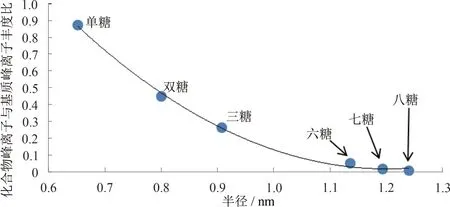
圖5 化合物峰離子與基質峰離子豐度比隨糖類化合物直徑的變化曲線Fig.5 Relationship between peak intensity ratio([M,analyte+Na]+ /[matrix+Na]+)and diameter of saccharides
3 結 論
將蒙脫石皂石黏土應用于MALDI-TOF-MS技術,將傳統的有機基質THAP 插入經陽離子交換后的皂石黏土晶面間隙中,制備成新型的復合基質,并將其應用于糖類化合物的檢測。由于受復合基質晶面間距的限制,只有小分子糖類化合物能進入晶面間隙充分接觸有機基質并被離子化,從而實現對小分子糖類化合物的選擇性檢測。
猜你喜歡
現代畜牧科技(2021年9期)2021-10-13 06:39:14
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
電子制作(2018年18期)2018-11-14 01:48:24
山東工業技術(2016年15期)2016-12-01 05:31:22
當代經濟研究(2016年5期)2016-12-01 03:12:05
現代農業(2016年5期)2016-02-28 18:42:46
出版與印刷(2016年3期)2016-02-02 01:20:11
中國中醫藥現代遠程教育(2014年11期)2014-08-08 13:23:44
華北水利水電大學學報(社會科學版)(2014年3期)2014-04-16 04:38:31
終身教育研究(2014年5期)2014-02-28 01:23:06
